
Carl Zimmer's Blog, page 34
January 17, 2013
The Sooty Greenhouse: My New Story For Yale Environment 360

Soot from a diesel-fueled truck. Image: EPA/ Wikipedia
The soot we loft into the sky is a remarkably mysterious player in the climate game. At Yale Environment 360, I report on the most comprehensive study yet of soot, which reveals that it’s trapping huge amounts of heat. Yet getting rid of all the soot we put in the atmosphere wouldn’t change the climate much. Check out my piece for the solution to that paradox.


Mutants: A Story About Tracking A Hospital Killer
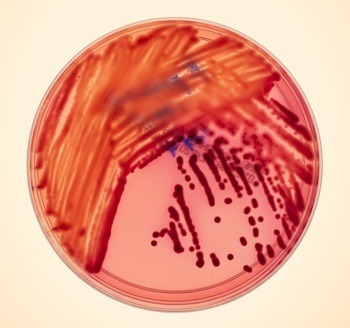
Klebsiella pneumoniae Photo: Dan Forbes/Wired
About 100,000 people die each year in the United States from infections they pick up in hospitals. Even the best hospitals in the country are not exempt from this disaster, and it’s getting worse: the bacteria that are attacking patients are becoming frighteningly resistant to antibiotics. Some are becoming resistant to everything doctors can throw at them.I recently went to Bethesda, Maryland, to visit doctors who struggled with one of these outbreaks at the NIH Clinical Center, one of the country’s premiere research hospitals. Most hospitals stay pretty quiet about their outbreaks, but the Clinical Center staff was far more transparent. They were willing to take me around the hospital as they described their struggles to stop the bacteria, called KPC, as it crept mysteriously from patient to patient and from ward to ward.
The story of the outbreak is pretty grim. Months after it seemed over, another patient died from a KPC infection, meaning that the bacteria still lurk in the hospital. But there’s one ray of hope. The doctors at the hospital joined forces with genome-sequencing experts, who probed the DNA of the bacteria for clues to its spread. This was one of the first times that genome sequencing helped–in real time–in the fight against a hospital outbreak. In the next issue of Wired, I chronicle this extraordinary piece of detective work. You can read my feature online here.
As I write in the story, genome sequencing could potentially help a lot in the broader war against hospital-acquired infections. But it won’t make much difference unless hospitals are united in using the best tools possible. When the Clinical Center doctors headed out to other hospitals to offer the lessons they had learned, they were startled to find that KPC was a fixture in many of the other hospitals. The very fact that this information came as a surprise to them shows just how little communication there is between hospitals about their outbreaks.
Julie Segre, one of the genome experts who battled the KPC outbreak at the Clinical Center, is convinced that patients and their families can help push hospitals to start using genome sequencing and other new tools to battle outbreaks.
“When a patient thinks about where to go for a stem cell transplant or a chemotherapy treatment, they think about who has a better medical team,” she told me. “But if you’re really making that decision, one of the things you need to think about is, who has the best infection control? Who’s in the middle of an outbreak? That’s what I’d want to know.”
To see what twenty-first century infection control looks like, check out my story.
January 16, 2013
The Hidden World Moves
Nikon has handed out awards in its second annual Small World In Motion competition–a contest for the best video of the world we can’t see with our naked eye.
Here are a few of my favorites…
A tiny animal called a rotifer lives inside a tube attached to a plant. It beats hairs called cilia to pump food into the tube, where it can be ground up by the rotifer’s jaw-like structures.
Here, kidney cells grow in a dish into the distinctive branched anatomy that lets kidneys filter waste out of blood.
Cells move by tearing down and rebuilding their internal skeleton. This video of a cell shows the filaments that make up the skeleton as glowing sticks. (Here’s an article I wrote about cell skeletons in the New York Times.)
The entire gallery of winners and runners-up is here.
January 14, 2013
Influenza: Our Incompetent Enemy
It’s not easy to push the explosive terror of norovirus out of the public square. It takes another virus that’s even more worrying and even more adept at spreading suffering around the world.
I speak, of course, of influenza.
This winter’s influenza season came early to the Northern Hemisphere and made a splashy debut. By the end of last week, the Centers for Disease Control reported widespread flu in 47 out of 50 states in the U.S. Some stocks of vaccines were running out. Some parts of the U.S. show signs of having hit their peak, but other places, like California, haven’t yet really experienced the full wallop of flu season. So we could be looking at several more weeks of serious trouble. Europe, meanwhile, is seeing high levels of flu infections, too. (I spoke today about the flu on KPCC–listen here.)
What makes all this all the more nerve-wracking is that influenza isn’t even bringing its A game right now. We’re not dealing with a new, super-virulent strain of influenza akin to the 1918 “Spanish flu,” which killed 50 million people worldwide. This is just regular seasonal flu–a stew of old-timer flu strains that slosh out of the tropics, heading north and south as conditions permit, and killing roughly half a million people every year. The fact that this year’s seasonal flu has overwhelmed us (shutting down some hospitals, in fact) doesn’t bode well for the inevitable moment some time in the future when we do come face to face with a much meaner flu strain.
The trouble lies not just in our creaky hospital system, but in the weapons we have on hand. There’s a flu vaccine, thank goodness, but it’s only about 62% effective. Making matters worse, well under half of Americans get vaccinated each year. If you get sick, doctors don’t have a lot of options for treatments. There are a couple antiviral drugs the CDC suggests to doctors, but it’s not clear how much (if anything) they do to alleviate symptoms. Making matters worse, the viruses are evolving resistance to those drugs. There are more anti-flu drugs in the pipeline, fortunately, but we’ll have to wait to see just how effective they are, and whether they will be wiped out by resistance as well. In October I wrote in the New York Times about new advances towards a universal flu vaccine that might provide a lifetime’s protection. While the research is heartening, it won’t help us for the next few years–if not the next few decades.
In the face of all these sobering facts, it’s easy to fall under the virus’s spell and be filled with awe. After all, with a dozen or so genes, it manages to replicate itself by the trillions and circle the globe in weeks.
But here’s the funny thing about influenza. It’s pathetic.
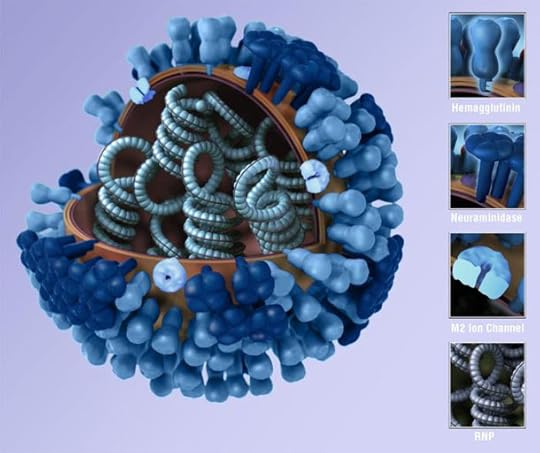
A team of scientists led by Jonathan Yewdell at the National Institute of Allergic and Infectious Diseases recently asked a simple question: what are the odds that an influenza virus is in good working order?
In order to infect a cell, flu viruses need to produce a protein called hemagglutinin, which forms knobs on the virus’s surface and latches onto host cells. Once the virus gets inside the cell, other proteins shut down the host cell’s natural antiviral defenses, help guide the cell to make new genes, and then help package them up. Another protein on the shell of flu viruses, called neuraminidase, opens up the host cell to allow the viruses to escape.
Yewdell and his colleagues infected cells with flu viruses and then surveyed them for these essential proteins. Time and again, they found viruses that lacked at least one of them. They tested out these defective viruses and found they failed to work as a virus should. As they report in an upcoming paper in the Journal of Virology, almost ninety percent are incapable of replicating because they’re missing at least one essential protein.
This is not some general feature of all viruses. Yewdell and his colleagues ran a similar experiment with another species, called vesicular stomatitis virus. It almost always managed to do the bare minimum and produce new viruses with all its essential proteins. Influenza stands out in its incompetence.
Yewdell and his colleagues offer up a few possible explanations for this incompetence.
–Influenza viruses mutate like mad. Mutations may be so common and so devastating that the vast majority of viruses end up unable to make at least one essential protein.
–It’s also possible that when a virus invades a host cell, it fails to get the cell to make all the essential proteins that new viruses will need. Nobody knows the success rate for this step of influenza infection is.
–Another possible source of failure has to do with the way genes are arranged in the flu virus. Rather than being one continue string of genetic material, the flu virus genome is broken up into eight segments. A new virus needs to swallow up a copy of all eight segments in order to have the genes for all its essential proteins. Flu viruses may be sloppy in assembling their genes.
It’s possible that all three causes are in play, or that influenza’s incompetence has yet another source. Whatever the reason, the simple fact remains: nine out of ten flu viruses are fundamental failures. Which raises the question of why there’s any flu at all.
Flu still manages to be a major threat to public health because viruses can handle failure very well. If only ten percent of viruses have all their essential proteins, they can carry on the flu virus legacy by replicating exponentially. And it may well be that the incompetent flu viruses can replicate as well. Two or more flu viruses sometimes infect the same cell at once. They may be able to help each other out, making up for the defects in their fellow viruses with their own working proteins. If one virus can’t protect itself against a cell’s defenses, another can. If one virus doesn’t have the proteins it needs to escape the host cell, another virus may open the door.
It’s almost sweet to imagine flu viruses helping each other overcome their collective weaknesses. Until, of course, that spirit of cooperation lands you in bed with a fever and the sort of aches you expect from a boxing match.
(For more information on flu and other viruses, see my book A Planet of Viruses)
Images: Doug Jordan, CDC
January 10, 2013
Watching Fish Climb Darwin’s Mountain
When biologists think about the evolution of life, they think about climbing mountains.
To understand their alpine frame of mind, imagine a biologist studying the fish in a lake. Each fish may be born big or small. Fish born at certain sizes may be more likely to survive and reproduce than others. Each fish may be aggressive or shy. Again, their aggressiveness may determine their odds of having babies.
To picture all of this, it’s very helpful to imagine a landscape. Each point on that landscape is a different combination of aggression and body size. They’re like the longitude and latitude on a map. Each combination leads to a particular level of reproductive success. Picture that success as the elevation of that point on the landscape. The more success, the higher the altitude.
The biology of those fish can give the landscape a topography. Perhaps it produces a single mountain. The peak is the combination of weight and aggression that produces the most possible babies. The landscape drops off in all directions, to combinations that make it more likely the fish will die, or fail to reproduce.
The actual fish in the actual lake might turn out to be clustered on one of the mountain’s flanks. The fish closer to the peak have more babies than the others further downhill. As a result, they’ll pass down more copies of their genes to the next generation. And that means that the population will climb up towards the peak. If a new mutation arises, natural selection will favor it if it helps the fish climb further. Eventually, the fish may reach the mountaintop. Once they plant their flag on the peak, they’ll be stuck. Natural selection won’t be able to nudge them off.
Now imagine that there are two peaks, not one. The fish sit on one mountaintop, while the second peak towers over them in the distance. They can’t get to that second peak, though, because natural selection can only nudge them uphill. They are stuck with a mediocre body.
This evolutionary landscape has floated in the minds of biologists for at least eighty years. A series of scientists, starting with Sewell Wright and George G. Simpson, developed the concept as a way to think about how evolution transforms populations. (For more information, check out the new book, The Adaptive Landscape in Evolutionary Biology.) For many biologists, it’s been a powerfully useful image. It’s even more useful if scientists can map landscapes with real biological data, setting the altitude at each point based on how well organisms really do reproduce.
Chris Martin and Peter Wainwright of the University of California at Davis have done just that, with real fish in real lakes. And their research is helping scientists to understand how evolution has allowed life to plant its flag not just on one mountain peak, but along entire mountain ranges.
The fish in question are three species of pupfish (Cyprinodon) that live in lakes on the island of San Salvador the Bahamas. The lakes are less than 10,000 years old, but they contain three species that feed in fundamentally different ways. One species eats algae and decomposing animal matter–a common lifestyle for pupfish found in other parts of the New World. But a second species is bizarre: it has powerful jaws that it uses to crush snails. And a third is even weirder: it chews off scales from other fish.
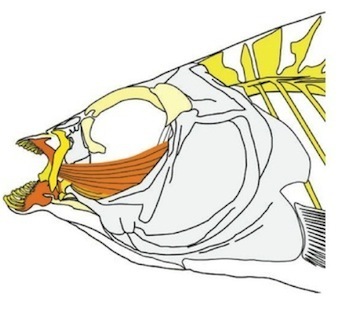
From Martin et al 2011
Just from an anatomical point of view, the emergence of these new forms was spectacular. This diagram shows the parts of the pupfish body that have changed on San Salvador (yellow shows strong changes, and orange is stronger). The changes are among the fastest ever recorded in animals.
To understand how the pupfish managed to evolve so quickly into three such distinct forms, Martin and Wainwright took advantage of the fact that the species are so young. They can still interbreed and produce hybrids. So Martin scooped up fish from the lakes and took them back to Davis, where he bred them for two generations to produce 3,000 hybrids, each with a different combination of the traits.
Martin studied some of the hybrids in his lab, and he found that they all survived about equally well there. Their success told Martin that combining their genes together didn’t create any disorders that cut their lives short. To see how they fared in the wild, he flew back to San Salvador with nearly 2,000 hybrid pupfish.
Martin dumped the fish into enclosures he built in the lakes. He let them fend for themselves for three months. Then he came back to see which ones were still alive, and how much the survivors had grown.
From this data, Martin could draw an accurate landscape of pupfish evolution. And here’s the landscape he drew. The latitude and the longitude represent two different sets of measurements, taking into account features such as the length of their jaw and the depth of their bodies.

The Adaptive Landscape of Pupfish. Courtesy of Chris Martin
The green peak is the shape found in the algae-detritus feeding species. The red peak represents the snail-crushing form. The intervening landscape is made up of valleys, reflecting the fact that the further away hybrids were from the original species, the worse they did. And down in the blue valley are the hybrids that resemble the scale scrapers.
Most of the San Salvador pupfish are algae-detritus feeders. But Martin’s research shows that if they could switch to feeding on snails, they’d be much better off. To do so, however, they would have to cross a deep evolutionary valley. As a result, they are stuck on their own peak, looking up at a better one.
So how did the snail-crushers evolve in the first place? One clue comes from another experiment Martin ran. He put the pupfish in another set of enclosures, but at much lower density than before. The lack of competition was good for all the fish, hybrids included; they were all much more likely to be alive when he came back to the enclosures. There was just more food to go around, and so being a clumsy hybrid didn’t impose much of a cost on a pupfish.
This result suggests that things were different in the lakes when they first formed thousands of years ago. There were no competitors when the pupfish arrived. In other words, the evolutionary landscape looked different. Instead of the Himalayas, think gently rolling hills. Some fish were able to start adapting to crushing snails. They were mediocre at first, but they were still the best snail-crushers on San Salvador. Their descendants evolved to become so good at it that they’ll outcompete any pupfish that might wander towards their own evolutionary mountain.
What’s happening in these lakes on one little island could shed light on how other species have evolved over hundreds of millions of years. Again and again, scientists find evidence that lineages explode into diversity quickly, and then their diversification tapers off. It seems that the adaptive landscape allows them to move fast to occupy empty niches. The landscape changes shape, making it much harder to move around it. Scientists who have documented these patterns of diversity in the fossil record have envisioned an evolutionary landscape with several peaks. Now, for the first time, Martin has marked those peaks in a real evolutionary landscape.
This experiment doesn’t answer all the questions scientists have about how diversity evolves, of course, and it even raises some questions of its own. What’s up with those scale scrapers, for example? Why are they down in the depths of the valleys? When Martin put hybrids with scraper-like anatomy in the lakes, they pretty much all died. What’s interesting is that Martin found few scales in their stomachs. It’s as if they didn’t even try to feed this way. It may be that scale eating demands such an extreme anatomy that Martin’s hybrids just didn’t have a chance of surviving in that manner. But they were also outcompeted by the other hybrids for the other food in the lakes, and so they died out.
In other words, even in these little lakes, there is an evolutionary terra incognita Martin has left to map.
[Mountains: Headlee Pass by yo tuco, Flickr/Creative Commons]
January 9, 2013
On The Origin of Venom
You are not venomous. Your spit, while unpleasant, cannot kill. Your dog is not venomous, either. Neither is the squirrel on the sidewalk, the bullfrog in the pond, or the dragonfly floating by your window.
Venom is the mark of a special club, a select subset of the animal kingdom. It includes vipers, jellyfish, spiders, scorpions, centipedes, bees, cone snails, newts, platypus, and even a primate. All these animals produce molecules that attack a victim in minutes or even seconds. The molecules are intricately complex, with shapes that allow them to precisely lock onto certain proteins on our own cells. Depending on its exact target in the body, venom will paralyze nerves, rot muscles, and stop blood from clotting.
Venomous animals are not all closely related to each other. The platypus is much more closely related to us, for example, than it is to a centipede. And that means that venom has independently evolved many times over in the history of life. First there were animals without venom, and then there were.
The origin of new adaptations is one of the most fascinating parts of evolution–from hands to feathers to eyes. But scientists have made exceptional progress in figuring out the origins of venom. Their success may have something to do with the fact that each type of venom is encoded by a single gene. That means that scientists can compare venoms gene by gene to see how they’re related to each other. That’s not to say that the venomous life is a simple one. After all, a rattlesnake produces not one venom but a cocktail of them; the cocktail in another snake will be profoundly different. Delivering the venom also requires a lot of equipment. Venom is produced in special glands, for example, and then has to get transported to the place where it will make contact with its victim–to the skin of a newt, to the tail of a scorpion, to the fang of a cobra. Nevertheless, the molecular elegance of venom lowers a ladder down to scientists, so that they can start their climb into the mysteries of venom evolution.
Recently, some of the world’s experts on venom came together to take stock of what they’ve learned. [pdf] The ways in which venoms emerge, and then evolve, offer lessons for the ways in which any complex new thing comes into being.
Venoms did not pop out of the void. They started out as genes for other functions. Venom genes are closely related to other genes that carry out entirely different jobs, both in venomous animals and non-venomous ones. Some venoms are closely related to immune system proteins, for example, which attack bacteria invading the body. Others are closely related to digestive enzymes
How does an enzyme end up as a venom? There are a number of ways. A common type of mutation causes DNA to get duplicated. At first, the duplication just means that twice as much of the original protein gets made. But then the extra gene can mutate again without harming the function of the original one. A mutation can, for example, change the signal a gene gets about where it should make its protein. Instead of becoming active in the pancreas, for example, it might start making proteins in the mouth.
When an animal bites its prey, the enzyme can then get into the wound. It might happen to have a harmful effect. Even a small effect could help the animal catch more prey, and thus be favored by natural selection. And the new proto-venom gene may undergo more evolution. It may become more and more toxic. Or it may duplicate, and the two venom genes may become deadly in different ways. Two genes can become four, four eight and on and on. The rate at which cone snails duplicate venom genes, scientists have found, is the fast gene duplication ever found in the animal kingdom.
Each new copy of a venom gene can get fine tuned even further. They may mutate a single base of their DNA, or they may undergo more dramatic changes. Genes are made up of segments called exons, and cells assemble the information in the exons in order to make a corresponding protein. In venom genes, exons sometimes get skipped or shuffled, producing new venom molecules with new properties. These new venoms allow animals to hunt new kinds of prey, or help them do a better job of killing old victims that evolve new defenses.
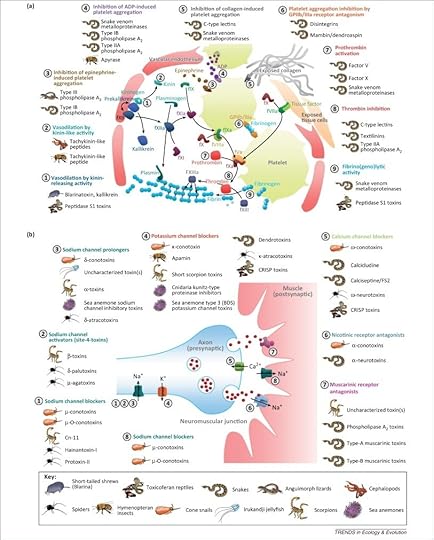
Each lineage of venomous animals became deadly on its own, independent of all the others. And yet, in the end, their venoms echo each other. I’ve reprinted a crazily detailed figure below, which you can enlarge by clicking on the image. It shows how different animals have ended up with venoms that zero in on the same molecular targets. The targets fall into two main categories: the channels and receptors on neurons, and the molecules involved in clotting blood. For example, cone snails, scorpions, and anemones have all evolved venoms that attack channels on neurons that pump out potassium. Snakes and bees have evolved the ability to block platelets from clumping together, a crucial step in blood clotting. These results show that there are a limited number of ways to kill your victim quickly. No matter what genes you borrow for the evolution of venom, they will end up very similar to other venoms.
There’s a lot left for scientists to discover about venom evolution. One of the most surprising recent discoveries is that the borrowing involved in venom evolution doesn’t go one way. Take the natriuretic venom found in snakes like the pit viper. It evolved from proteins found in other animals (including us) which relax the walls of blood vessels. In snakes, it evolved into a venom that relaxes blood walls so fast that it causes a debilitatingly rapid drop in blood pressure. But in pit vipers, one of these natriuretic venoms is produced inside their brain.
No one knows what this venom is doing inside the snake’s brain. But it’s obvious what it’s not doing: killing prey. It’s likely that the venom, borrowed from other parts of the body, has now been borrowed back. That’s one of the most interesting evolutionary lessons that venom has to offer: it’s endlessly creative, yet remarkably frugal.
Photo by martinbuck119 via Flickr
January 7, 2013
If You’re Going To Live Inside A Zombie, Keep It Clean
If you haven’t already met Ampulex compressa, otherwise known as the jewel wasp, now is as good a time as any. Someday you may be very grateful that you did.

Ampulex compressa. Photo by K. Seltmann, via Creative Commons. Link: http://www.morphbank.net/?id=102143
This gorgeous animal, which measures just under an inch from mandibles to tail, lives across much of Africa and Asia, as well as a few Pacific Islands. Don’t be fooled by its lovely glittering appearance, though. This is a deeply sinister creature. Jewel wasps don’t rear their young in a familiar paper nest. For them, home is the inside of a cockroach.
When the female wasps are ready to lay their eggs, they take to the air and search for roaches. They find them on trees, on the ground, and even in people’s apartments. Since cockroaches don’t want to play host to their young, the wasps have to sneak up on their victims and subdue them–without killing them. So the wasp sneaks up and clamps its mandibles on the roach. As the roach tries to shake it off, the wasp hooks its tail underneath and stings its victim just below the head, temporarily paralyzing the roach’s front legs. Now the roach is easier to handle. The wasp then conducts brain surgery.

Ampulex compressa injects drugs in the brain of a roach. Photo by Ram Gal
As I’ve written about in greater detail here and here, the jewel wasp snakes its stinger up into the cockroach’s brain, using sensors at its tip to feel its way to specific regions where it then releases cocktails of neurotransmitters. The wasp removes her stinger and walks away to find a crevice that will serve as a suitable burrow. Her first sting wears off, and the roach is now free to run away. Except it doesn’t. It becomes the insect equivalent of a zombie, having lost all will.
The wasp returns–sometimes as long as half an hour later–and bites off one of the roach’s antenna. She slurps some roach juice from the wound, like a kid drinking a milkshake through a straw. Then she bites down on the antenna stump and guides the roach to her burrow, leading it as if it were a dog on a leash. The roach goes without a fight. The wasp leads the roach into the burrow and then lays an egg, shaped like a grain of rice, on its underside. Then she leaves the burrow, sealing it up to leave the roach in darkness.
The egg hatches, and out comes a larva. It chews a hole into the roach’s underside, from which it feeds. It grows larger and larger over the course of a week. And then it crawls inside.
Remember, the roach is still healthy. It could push away the larva, bolt for the exit, push aside the pebbles blocking the burrow, and scramble away. But it can’t, or it won’t–I’ll leave it to philosophers to find the right language to describe the free will of cockroaches. It just stands where it was led, while the wasp larva feeds on its insides and grows to fill the roach’s entire body cavity. Eventually the roach dies, and inside the cadaver, the wasp spins a coccoon around itself, inside of which it grows eyes and legs and wings. And then it pops out, ready to meet the world. The wasp shakes itself off, climbs out of the burrow, dries out its wings, and flies away. It leaves behind the dead husk that was the roach.
 There’s a world of adaptations that the jewel wasp deploys in order to turn this science fiction story into natural history. Many of those adaptations have to do with brains: how the wasp brain manages to carry out its complex stinging maneuver, and what exactly it does to its victim’s brain to make it a zombie. But scientists are also discovering adaptations of an entirely different sort. In order for the jewel wasp to make roaches its home, it has to be more than a brain surgeon. It also has to be a pharmacologist.
There’s a world of adaptations that the jewel wasp deploys in order to turn this science fiction story into natural history. Many of those adaptations have to do with brains: how the wasp brain manages to carry out its complex stinging maneuver, and what exactly it does to its victim’s brain to make it a zombie. But scientists are also discovering adaptations of an entirely different sort. In order for the jewel wasp to make roaches its home, it has to be more than a brain surgeon. It also has to be a pharmacologist.
Gudrun Herzner, a biologist at the University of Regensburg in Germany, was curious about how the larvae manage to stay alive inside a cockroach. To a jewel wasp, the inside of a roach may be an all-night diner but it’s also a hot zone. Cockroaches carry plenty of microbes–including antibiotic-resistant pathogens that are the bane of hospitals and nursing homes. The pathogens aren’t just dangerous to humans, though. When Herzner looked inside roaches, she found them to be packed with a microbe called Serratia that’s both a medical menace and a threat to insect larvae. If the inside of a cockroach is an all-night diner, it’s a diner where the short-order cook douses all the orders with deadly bacteria before the waitress delivers the dishes.
Obviously the wasps are not dying inside their roach hosts, even after their hosts die and presumably start to rot. So they presumably have some kind of defense against infection. To see what that defense might be, Herzner created a hole on the side of infected roaches, which she covered with a little window. She then spied on the wasp larvae inside.
Herzner saw something no one had seen before. The larvae produced clear droplets in their mouths, which they carefully applied to the inner walls of the cockroach’s body cavity. The wasps then crawled around the cavity, smearing the droplets smoothly across all the inner surfaces of their host.
Fortunately for Herzner, the wasps also applied some of the drops to the window. She was able to extract chemicals from the drops, which she then analyzed. Once she had identified those compounds, she gathered more of them by grinding up infected roaches.
She found a number of molecules in the droplets, two of which were especially interesting. They’re called mellein and micromolide. And Herzner found that they stopped the growth of Serratia she isolated from the cockroaches, along with other pathogens. When the wasp enters its cockroach diner, in other words, it sponges down the walls, the tables, the dishes, and the food with an antibiotic soap.
In my book Parasite Rex, I wrote about how parasites long had a reputation as degenerates. They evolved from free-living species by losing the adaptations necessary for a rugged life of self-reliance, trading their autonomy for a life as a moocher. This is merely a case of our imposing our own social anxieties on nature. In fact, parasites have to gain many adaptations in order to enter the altnerate universe that exists inside living hosts. It’s true that for a wasp larvae, it’s great to grow up inside a roach where your next meal is one slurp away. But it also means that you’re check-by-jowl with bacteria that could kill you.
The antibiotics that the jewel wasp has evolved are not entirely new. Scientists have found other forms of mellein in fungi, for example. Micromolide was first discovered in a citrus plant called Micromelum hirsutum. The wasps, in other words, have unwittingly hit on the same solution for fighting off bacteria that radically different organisms did on their own.
The jewel wasp is worth getting to know just because it exists. But now it’s possible that we might someday benefit from that knowledge in the most practical way imaginable. What is good for jewel wasps might prove good for us.
The mellein found in fungi is effective against MRSA, the deadly strain of skin bacteria that is resistant to most antibiotics. When scientists tested out the micromolide from plants on the bacteria that causes tuberculosis, it proved to be among the most powerful anti-TB drugs ever found. Now the jewel wasp turns out to be a factory for similar antibiotics, which might turn out to be even better than what’s been found in fungi or plants. To Herzner, that possibility cries out for exploration, because right now, the antibiotics we use to cure our own infections are failing at a distressing rate. “I personally believe that we no have no other choice but to look for alternatives to the commonly used antibiotics,” says Herzner.
Of course, just finding a natural antibiotic does not mean that tomorrow we can buy it in pill form, as Ed Yong recently reminded us. But the first step on the long road to replenishing the antibiotic cabinet is to start looking in unexpected places. Places like zombie cockroaches.
Reference: Herzner et al, 2013. Larvae of the parasitoid wasp Ampulex compressa sanitize their host, the American cockroach, with a blend of antimicrobials. PNAS, to be published the week of 1/7/13. When the paper is published, you’ll find it here.
[Update 1/7 7 pm: Herzner's quote fixed]
January 6, 2013
How Quarks Changed A Life (Science Tattoo Sunday)
 Xue Sun writes, “I’ve attached a photo of some relatively recent ink I got on my waist, done by Nalla at Goose in Brooklyn, NY. It’s a diagram of the bubble chamber event that led to the discovery of the charmed baryon. While I’m actually a neuroscientist and not a physicist, it was learning about quarks in high school that gave me my first ‘Wow! Science!’ moment. When I found this image, I immediately knew I had to have it as a tattoo.”
Xue Sun writes, “I’ve attached a photo of some relatively recent ink I got on my waist, done by Nalla at Goose in Brooklyn, NY. It’s a diagram of the bubble chamber event that led to the discovery of the charmed baryon. While I’m actually a neuroscientist and not a physicist, it was learning about quarks in high school that gave me my first ‘Wow! Science!’ moment. When I found this image, I immediately knew I had to have it as a tattoo.”
Some details about the discovery of charmed baryons, plus the original diagram, can be found here.
You can see the rest of the Science Tattoo Emporium here and in Science Ink: Tattoos of the Science Obsessed.
January 3, 2013
Life Under A Faint Sun
If you could have looked up at the sky 4 billion years ago, you would have seen a sun much dimmer than ours today. And if you looked down at the Earth’s oceans, you would have seen an expanse of bobbing waves.
That’s a problem–a simple one, but a big one. And scientists have been wrestling with it for fifty years.
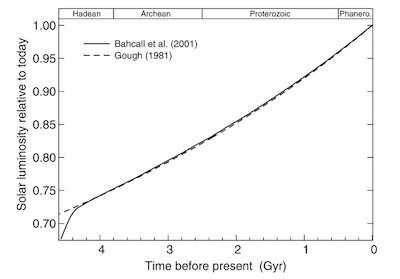
The brightness of the sun over the past 4.5 billion years. From Feulner 2011 http://arxiv.org/abs/1204.4449
The evidence for these two facts about the early Earth–a dim sun and liquid oceans–were already strong in the 1960s. Astronomers have compared our sun today to other stars of different sizes and ages, and they’ve been able to reconstruct much of its history. The sun started out about 70% as bright as today. It slowly grew brighter; even two billion years ago (2.5 billion years after the Earth formed), the sun was still just 85% as bright as today.
On its own, the faint young sun could not have kept the Earth from freezing over. And yet there are lots of signs in ancient rocks that the Earth was wet. Tiny crystals dating back over 4 billion years have a chemistry that required liquid water. Ancient rocks known as pillow lavas must have formed as molten Earth oozed out into sea water.
As early as the mid-1960s [pdf], scientists realized that these two lines of evidence posed a paradox: what is now known as the Faint Young Sun Paradox. It was a serious problem that required serious thought. It didn’t just mean that the evidence from geology and astronomy wasn’t meshing together. It also added a puzzle to the rise of life on Earth. Life would have had a hard time getting started on a planet of ice.
In 1972, Carl Sagan and his colleague at Cornell George Mullen proposed a solution to the paradox: the greenhouse effect. When radiation from the sun hits the Earth, some of it bounces back into space, but some of it lingers, thanks to heat-trapping gases in the atmosphere. The early Earth would have released gasses from its rocks, creating the first atmosphere. If it had the right chemistry, Sagan and Mullen argued, it might have been able to keep the Earth warm enough to melt ice. They suggested ammonia as a plausible heat-trapper on the young planet.
Unfortunately, ammonia turned out to be a bad solution. Other scientists figured out that ultraviolet rays from the sun would have destroyed any built up ammonia in the atmosphere in less than a decade. That’s not much of a defense against the deep freeze.
But ammonia is not the only greenhouse gas in the game. Today, carbon dioxide and methane are two important molecules keeping our planet warm (and warmer). Scientists have tried for years to narrow down the possible range of the two gases on the early Earth. It’s a very tricky puzzle, because scientists know that there are many factors that can influence their concentrations. And there were factors on the early Earth that we don’t experience today, such as a fairly steady bombardment of comets and giant meteors. Making matters even more complicated, greenhouse gases are not always greenhouse gases. Once the proportion of methane to carbon dioxide gets too high, it produces an organic haze that bounces radiation back into space, cooling the planet.
The consensus today is that methane and carbon dioxide may have warmed the early Earth a fair amount, but not enough to solve the paradox. So scientists are looking at other possible factors. Clouds may have helped. The early Earth rotated quickly through a 14 hour day, which may have changed how the oceans circulated–and thus how they trapped heat. But wide scope still remains for more ideas.
Today in Science, Robin Wordsworth and Raymond Pierrehumbert of the University of Chicago offer two new players to the Faint Young Sun game. They argue that a pair of molecules that have hitherto been neglected–hydrogen (H2) and nitrogen (N2)–could have made up a lot of the difference between the sun’s feeble glow and the Earth’s life-sustaining warmth.
There’s hardly any molecular hydrogen in our atmosphere today, because it easily skips out of the atmosphere into space. But Wordsworth and Pierrehumbert argue that such an escape would have been much harder for hydrogen on the early Earth, partly because it couldn’t get as big of a boost from ultraviolet rays from the sun. They estimate that hydrogen could have made up as much as thirty percent of the atmosphere. They also argue that nitrogen levels were three times higher than today.
On their own, nitrogen and hydrogen don’t do a very good job of trapping the sun’s heat. But when they crash into each other, their structure briefly changes, allowing them to absorb radiation. Wordsworth and Pierrehumbert built a model of a hydrogen and nitrogen-rich early atmosphere and found that as the molecules crashed into each other, they soaked up a lot of heat–enough, they argue, to heat the planet 10 to 15 degrees centigrade. That would go a long way to resolving the Faint Young Sun Paradox.
This new study probably won’t bring the fifty-year debate to a halt. In an accompanying commentary, James Kasting at Penn State argues that nitrogen is too heavy to absorb much radiation, even in the midst of a collision. Instead, Wordsworth and Pierrehumbert are stocking the cabinet that aspiring chefs can raid when they are trying to come up with new recipes for the early Earth.
If hydrogen and nitrogen do turn out to be part of the answer to the Faint Young Sun Paradox, they may have some fascinating implications about life on Earth–and elsewhere. Molecular hydrogen is fine dining for certain types of microbes known as methanogens. As soon as they evolved, they would have been able to feast on a sky full of hydrogen. By devouring the Earth’s protective hydrogen, they might have cooled the planet until it experienced its first glaciers. And beyond Earth, we may need to expand our concept of what kind of planet could support life. If they turn out to have a rich supply of hydrogen and nitrogen, they may offer a toasty incubator for aliens.
Image: The “Fighting Temeraire” Tugged to her Last Berth to be Broken up via Wiki-Paintings
January 2, 2013
The Norovirus: A Study in Puked Perfection
Today, The Guardian relayed one of those stunning medical stories that causes me to clean off my glasses and take another look to make sure I’m reading it clearly. They report that an outbreak of norovirus in Britain this winter has struck more than 1.1 million people with vomiting and diarrhea.
That’s right: 1.1 million. In Britain alone.
What is this fearsome bug, you may be asking, and why isn’t it the subject of a Hollywood horror movie?
Noroviruses are one of virology’s great open secrets. In a recent issue of The Journal of Infectious Diseases, Aron Hall of the Centers for Disease Control declared, “Noroviruses are perhaps the perfect human pathogen.”
Here’s what inspires awe in scientists like Hall.
Each norovirus carries just nine protein-coding genes (you have about 20,000). Even with that skimpy genetic toolkit, noroviruses can break the locks on our cells, slip in, and hack our own DNA to make new noroviruses. The details of this invasion are sketchy, alas, because scientists haven’t figured out a good way to rear noroviruses in human cells in their labs. It’s not even clear exactly which type of cell they invade once they reach the gut. Regardless of the type, they clearly know how to exploit their hosts. Noroviruses come roaring out of the infected cells in vast numbers. And then they come roaring out of the body. Within a day of infection, noroviruses have rewired our digestive system so that stuff comes flying out from both ends.
To trigger diarrhea, the viruses alter the intestinal lining, causing cells to dump out their fluids, which then gets washed out of the body–along with many, many, many noroviruses. Each gram of feces contains around five billion noroviruses. (Yes, billion.)
Noroviruses also make us puke. And if you can gather enough strength to think clearly about this, virus-driven vomit is a pretty remarkable manipulation of a host. Vomiting occurs when our nerves send signals that swiftly contract the muscles lining the stomach. Vomiting does us a lot of good when we’re hurling out some noxious substance that would do us harm. But repeated projectile vomiting of the sort that noroviruses cause serve another function: they let the viruses to find a new host.
To get us to throw up so violently, noroviruses must tap into our nervous systems, but it’s not clear how they do so. Here’s one particularly creepy hint: some studies indicate that during a norovirus infection, our stomachs slow down the passage of food into the intestines. In other words, they seem to load up the stomach in preparation for vomiting. Every particle of that stored food is a potential vehicle for noroviruses when it comes flying out of the mouth.
Once the norovirus emerges from its miserable host, it has to survive in the environment. Noroviruses have no trouble doing so, it seems. Fine droplets released from sick people can float through the air and settle on food, on countertops, in swimming pools. They can survive freezing and heating and bleaching. In 2010, scientists surveyed a hospital for noroviruses and found 21 different types sitting on a single countertop. It takes fewer than twenty noroviruses slipping into a person’s mouth to start a new infection.
This natural history makes for Olympic-level feats of transmission. In 2010, for example, nine members of a girl’s soccer team got sick with noroviruses while on a trip for a tournament in Oregon. The outbreak began with one girl coming down with stomach pains one Saturday evening. She moved from her hotel room to stay with a chaperone, where she then had diarrhea and vomited through the night. The chaperone took her back home in the morning (and also became sick later). Only on Tuesday did the rest of her team get sick.
Epidemiologists figured out that the first step in the transmission took place in the chaperone’s hotel room. There was a reusable grocery bag sitting in the bathroom–which the first girl never touched as she went in and out through the night. The next day, another chaperone got the bag and brought it to another hotel room for lunch. It contained sealed containers of chips, cookies, and grapes. Seven of the eleven people who ate that food got sick.
Another display of the norovirus’s tenacity came with a study of a New Zealand plane in which an infected passenger threw up on the floor of the economy section. A flight attendant cleaned up the mess, and over the next week, the plane continued to fly without any cases of vomiting. Nevertheless, the norovirus managed to infect new hosts. Out of 63 flight attendants who worked in the plane over the next six days, 29 got sick–an attack rate of 42.9%.
No one can say how the current outbreak in Britain got its start, but its timing is typical: January is peak norovirus season. Places where people are in close quarters are especially good incubators for the virus. The Queen Mary II, for example, is currently getting scrubbed down after a bad outbreak. But cruise ships are hardly the only place where noroviruses thrive. Schools get cleared out from time to time by the pathogens (the name norovirus comes from Norwalk, Ohio, where it was first isolated from a school during a 1968 outbreak). Nursing homes are fertile ground, too, in part because people there often have weak immune systems. While healthy people can clear out a norovirus after a couple of exhausting days, the virus can cling to people with weak immune systems for months or even years.
Noroviruses are so good at spreading that it’s quite likely that at some point in your life, you’ve had a norovirus infection. (You may have wrongly called it a stomach flu. Flu–short for influenza–only infects people’s airways.) It’s estimated that in the United States alone, noroviruses infect at least 23 million people a year. Seventy thousand of them end up hospitalized, and nearly 800 die. Things are worse in developing countries, where people are less likely to get rigged to an IV to get pumped full of fluids. It’s estimated that noroviruses kill 200,000 children under the age of five every year in developing countries.
It would be very nice if we only had to worry about getting noroviruses once and then could enjoy protection from them for the rest of our lives. Unfortunately, it seems that we only have a brief protection of perhaps a few months, and then we’re fair game again. As a strain of norovirus encounters this short-lived defense, it evolves new ways to evade our immune systems. A modified strain can then sweep around the world in as little as three months.
While some drugs show promise in blocking noroviruses from infecting cells, none have passed muster in a clinical trial on people. The best hope to put a real dent in the reign of noroviruses may be vaccines. Last year, scientists at Baylor College of Medicine reported that a vaccine could provide some protection against the virus. When people were exposed to noroviruses, 69% of people who got the placebo became sick, compared to only 37% of the vaccinated subjects.
A vaccine that leaves more than a third of people vulnerable to a virus is not exactly a silver bullet. But against such a perfect pathogen, even a little relief can ease a lot of pain.
For more information, check out norovirus expert Stephanie Karst talking about noroviruses with the gang at This Week in Virology. For viruses in general, see my book A Planet of Viruses
[Image: German Cancer Research Center]
(Update 5:30 pm: Changed England to Britain and fixed some typos. 7:30 am: corrected Norfolk to Norwalk, clarified timing of British outbreak)


