
Mark S. Senak's Blog, page 8
March 10, 2020
OPDP Issues First Letter of the Year

Hey! 2009 called. They want their Warning Letter back!
Last week FDA’s Office of Prescription Drug Promotion (OPDP) posted the first letter issued for 2020 that was sent in February for the most common violation of all – related to the minimization or omission of risk information. This evoked shades of April 2009 when the agency issued letters covering 45 brands to 14 companies for banner ads on the Internet. Only a month before OPDP had said that the guiding rule of thumb in regulatory enforcement in digital media was that they were concerned with the “message not the medium”. Then they issued a slew of notice of violation (NOV) letters aimed at a medium.
But the reason for those letters, and this one, was one that was indeed about the message. It lacked risk information on its face and presumably it was offered at the landing site to which the sponsored link took one who clicked on it.
Even though the letter involves and age-old regulatory issue – the inclusion of risk information – and a set of facts we have seen before, there are some things to take away from this particular letter. Not only did the agency take regulatory action on this promotion, but it issued a Warning Letter (WL) rather than a Notice of Violation (NOV) a/k/a Untitled Letter. What are the insights from this action? I have no line into OPDP, but here are a few independent observations:
First of all, in this instance, the promotional communication was in connection with a product that also has a boxed warning on the label. The presence of a boxed warning indicates a special concern regarding the use of the product that is being emphasized. It is also a surrogate marker for those responsible for promotional communications to take extra special care. In this case, there were safety issues related not only to a potential for abuse (an area of specific concern overall) to the potential for cardiovascular adverse events if misused or used by patients with specific conditions.Another factor is that the product is a treatment for ADHD – a treatment category which has had (by my count from my database) 13 letters issued since 2005, 9 of which involved the exclusion or minimization of risk information and 4 of which involved the use of a WL over NOV. If you are responsible for communications for a product, it is not a bad idea to get an idea of the causes of regulatory actions by OPDP in the past – what were the tripwires and lessons to be learned?And there is the fact that the advertisement was directed to the use of the product with children. When dealing with a vulnerable population it is important to take extra care in communications to ensure that the potential for a violation is utterly minimized. Finally, the letter refers to complaints from the “Bad Ad” program, indicating that this effort of OPDP to deputize health care professionals to be alert and inform FDA of potential violations is still active.
Enforcement has been at a very low rate for a long time, but low enforcement does not mean no enforcement and the agency has said that it is turning its focus to communications which pose a higher level of concern such as risk information lacking for products with a higher level of risk profile and for pre-approval promotion. Every promotional communication requires scrutiny, but when a product has some specific traits – a category that is scrutinized, a vulnerable population and/or a boxed warning – these are characteristics that merit heightened regard.
Photo by Goh Rhy Yan on Unsplash
Share this:







March 6, 2020
Meetings, Clinical Trials and Supply Chain Disruption from a Virus We Never Heard of Until New Year’s Eve…

The world changes quickly. A few weeks ago, I published an article on LinkedIn “Communications Challenges for Medical Manufacturers as COVID-19 Epidemic Emerges”. It was meant to outline some thinking for the potential for the disruption of medical meetings, of clinical trials and of course, the much talked about potential for supply chain disruption.
We are no longer talking about potential. In the few weeks since then, the European Society for Radiology which was to have its annual meeting starting next week, postponed the meeting and HIMMS 2020 was also cancelled. At least one pharmaceutical company, one which by the way got FDA approval for its multiple myeloma product just last month, has indicated that sales will be impacted and clinical trials may be impacted which would in turn, have a negative effect on the bottom line. And the government of India announced that the export of 26 pharmaceutical ingredients would be banned for export, likely to have a direct effect on the production of some specific categories of medicines including generic medications and over-the-counter pain relief.
That raises many problems for pharmaceutical and biotech companies. Of those, let’s concentrate on three:
Meeting Cancellations – Companies will have data milestones in the coming months with the possibility that the traditional venue for unveiling and discussing such milestones – i.e., the medical meeting – may either be cancelled or attendance may be diminished. Medical meetings are a primary avenue of communications for the exchange of scientific information regarding new therapies and understanding the latest developments in specific therapeutic categories;Media Environment – Companies will be dealing with a media environment that is not particularly interested in new therapies, unless they happen to apply to COVID-19. A colleague of mine wrote an article, also on LinkedIn, that took a look at coverage in the New York Times over the past few months and he makes some solid recommendations. The viral pandemic (let’s call it what it is) is grabbing headlines, ink and reporter attention;Tough Questions – Companies that are lucky enough to be engaged with media during this time are likely to face questions and scrutiny over supply chain continuity, and to a lesser degree, clinical trial timelines and of course, profitability.
There are no magic bullets. But there is strategic thinking. Here is are a few thought starters related to the above considerations:
Direct to Audience Communication – It is time to brainstorm over ways that companies can achieve at least some of the communications goals that would be associated with large medical meetings by employing digital means for conveying messages. If there was to be a poster presentation for a meeting, put it on video and make it available. In the absence of a meeting venue, develop and conduct webinars to cover presentations that were to occur. Think of every way possible to go virtual and execute upon it, then get the contents pushed out to patient groups, members of professional societies and investors. Focus on Trade Media – With COVID-19 taking up all of the ink in mainstreams, there is going to be a diminished capacity – and even a diminished interest by reporters – for non-COVID-19 news and data milestones – particularly those for non-life-threatening conditions – may get far less traction (or even none) by traditional means. For trade media, however, it is still their primary focus. Shift primary focus to these outlets and utilize bloggers and target online influencers more heavily and skillfully.Plan, Plan, Plan. The basis of planning is information and the situation is rapidly changing and doing so hourly. It is extremely important to know what is going on with respect to the numbers and the developments – both the medical and the policy ones – on an on-going basis and to consider their impact on one’s own operations. As the organizers of medical meetings grapple with whether or not to hold a meeting, individual medical manufacturing companies are imposing their own restrictions on employee travel and meetings. While data milestones are going to take a back seat, media scrutiny on supply chain flow will heighten. Scenario planning and messaging around worst case scenarios that considers both health implications and corporate reputation needs to be robust and recalibrated almost daily. Some pharmaceutical companies have reassured concerns about supply chain flow – but the situation on the ground can change quickly. Planning needs to be extensive.
For many, it is important to feel ahead of the curve. Under the current circumstances, there is no real curve as we catch up to determine the real extent of the outbreak and then can assess its real impact. There are many uncertainties – but one thing that is certain – traditional communications approaches are likely to require adjustment.
Photo Source: C.S. Goldsmith and A. Tamin
Share this:
February 21, 2020
Emerging Pathogens, Communications

In June 1981 I graduated from law school. One month after that an article appeared in the New York Times with the headline “Rare Cancer Seen in 41 Homosexuals” on page A20. These two events would end up pretty much defining the rest of my life. I became the chairperson of a group of lawyers volunteering time to provide legal help to people with this new disease and later became the Director of Legal Services at the Gay Men’s Health Crisis (GMHC) where I provided wills and advocacy on insurance and immigration matters, landlord/tenant issues, and discrimination in jobs and public accommodation. I also emerged as the spokesperson for the agency on important public health policy matters both at GMHC and in the 1990s at AIDS Project Los Angeles. This included commenting on important milestones such as actions by legislatures (or lack thereof), regulatory matters, and high profile developments such as Rock Hudson, Greg Louganis, Brad Davis and Magic Johnson, and more. In short, I got to know media and communications in the face of an emerging pathogen. During past outbreaks of emerging pathogens, I have written postings on this topic. As I watch the media report on the new Wuhan coronavirus, there are some lessons from that time that bear noting and repeating.
Facts are low; speculation is high – In the earliest reporting on Wuhan, human to human transmission was regarded as a low likelihood though transmission is now regarded as more likely than either SARS or MERS. So little in the earliest stages of an emerging situation that there is necessarily a good deal of speculation, often on which opinion gets based. Early reporting referenced the fact that mortality seemed concentrated in the elderly with co-morbidities, but news outlets also reported the deaths of young medical workers. In short, speculation or casual observation is often presented as fact and policy actions are sometimes based on that. But the only fact is that we don’t know much about this and only time and experience will tell. Numbers don’t mean a lot – The number of Wuhan cases reported climbed dramatically within only a few days. In the early days of an epidemic, numbers are frequently subject to change. The number of diagnoses represent only those people infected who are showing symptoms, not the number of people infected who may not be symptomatic but who may be able to transmit. Second, the method and nature of counting (and testing) can change, as it did for COVID-19 when the methodology for counting cases caused a spike in the numbers. Third, there is always under-reporting. A communications focus should rely less on numbers than on solid principles of public health and on the business facts that are at hand. Numbers are indicative, but not certain. Points of reference will change – In the earliest days of the AIDS epidemic the disease was referred to as GRIDS (Gay-related Immune Deficiency Syndrome) and then AIDS. When finally discovered, the virus was HTLV-III and then became called HIV. In short, it takes time for the nomenclature and framework to become established, which can add to confusion. The situation can remain very fluid well into an epidemic. The coronavirus became COVID-19. It will be essential to monitor and educate stakeholders, both internally and externally, in any organization. Fraud potential is high – Not only will homespun remedies emerge, but on a more unpleasant note, there is a high potential for products to appear accompanied by unapproved claims. There is also a high potential for the emergence of counterfeit products. This is where FDA and FTC have a role to play. Policy can ham-fisted – With so few facts in hand, policymakers often react in ways to make it appear that something is being done to address an outbreak when in fact, policies being enacted may not support public health goals, necessitating vigilance in monitoring and considerations for how organizations respond. Some may be benign while others may be truly ill-advised.
Under fast changing circumstances, it is difficult to get a good grasp of the total picture along with all the nuance represented therein. As events unfold, and there is a greater demand for communications from stakeholders such as public officials, companies and health authorities, it is important to keep in mind some truisms during the early days to help us keep our bearings and develop perspectives.
Information Resources
FDA Web Page – Outlines FDA actions with respect to COVID-19 and other important linksWorld Health Organization – Multiple resources and up-to-date situation reports with a focus that is both global and localizedCenters for Disease Control – Comprehensive overview of resources and information page on COVID-19 in the U.S.
Photo credit – NIAID-RML
Share this:
February 13, 2020
The Side Effects of a Coronavirus Epidemic for Pharma

There are a lot of important facts on the FDA’s Web Page set up to provide information about the novel coronoavirus (2019-nCOV) outbreak, now dubbed COVID-19. But there are three sobering statements that stand out:
There are no FDA-approved diagnostics for COVID-19There are no FDA-approved vaccines to prevent COVID-19There are no FDA-approved drugs to treat COVID-19
Statistics about the numbers of people who have been infected vary greatly. It is far too early to tell what will happen with this outbreak. It is not to early to prepare for it. Without the ability to readily diagnose, prevent or treat the condition – things that cannot be readily done – the main thing that can be done is planning.
When we think of an epidemic, we naturally think of the obvious concern – the safety of ourselves and those we care about. But there are consequences in store beyond that for many industries – most obviously the travel industry. And there are also dynamic concerns for the industry that makes our medicines.
API – First there is the potential for disruption of the production of medicine due to a lack of active pharmaceutical ingredients (API), a large portion of which are manufactured in China. While there are non-Chinese API producers, many of them are in China and it stands to reason that disruption is a distinct possibility. That would have an impact on everyone relying on medicine not only to treat infections, but also to treat chronic conditions such as diabetes, blood pressure and cholesterol levels. Manufacturing Disruption – In the event of a widely spread contagion, it stands to reason that social isolation practices – i.e., not coming to work – could become necessary and thereby interrupting the actual manufacture of finished medicines. While API can be obtained from various sources, the manufacture of many medicines is specific to established sites.Medical Meetings – The effects of the outbreak on large meetings is already becoming apparent with high-profile companies pulling out of the Mobile World Congress set for Barcelona this month resulting in the cancellation of the meeting. At the very time when collaboration is essential, there may be a disruption to medical meetings commonly held around the world at which clinical data is presented and cutting edge research is discussed. A disruption in the course of research-related discourse could impact the speed of development of new medicines. Regulatory Oversight – Media reports this week mentioned that visitors to the campus of the Food and Drug Administration were being asked upon entry about foreign travel. Social distancing also means that regulatory meetings such as FDA Advisory Committee meetings may be put on hold – as well as the review process itself – considerably slowing down the approval of new medicines. Blood Supply – As anyone connected with the early days of the AIDS epidemic can attest, when you have a communicable disease where there are no diagnostics for screening, the blood supply becomes particularly vulnerable, resulting in the potential for severe shortages to emerge.
These concerns are not new with COVID-19. The circumstances were very similar during the Avian flu outbreak during the first decade of the 2000’s and extensive planning around the supply chain was undertaken by the pharmaceutical industry. We find ourselves again with an imperative to not just plan appropriately but to consider how to effectively, accurately and compassionately communicate what needs to be considered in a time of crisis. In that way, industry can minimize some of the potential side effects that are possible as we see our way through this.
The World Health Organization puts out daily situation reports that are highly detailed and comprehensive. You can access them here.
Photo – Centers for Disease Control
Share this:
February 12, 2020
FDA Takes A Number of Actions to Enhance the Market for Biosimilars – A Process Not an Event

There is a policy interest in driving the biosimilar market. Among other reasons, increasing the number of generic drugs and biosimilars on the market addresses another policy priority – having an impact on the high cost of medicine. The approval of generic drugs has been at record highs the past few years, and FDA is also doing all that it can to facilitate the market for biosimilars. This is one of the few things FDA can actually do to address the issue of price, even if in the totality of things it is only nibbling away at the edge of a much larger issue.
So far in 2020, FDA has taken a series of actions designed to enhance the market for biosimilars.
On February 3, FDA posted a draft guidance document for public comment entitled “Biosimilars and Interchangeable Biosimilars; Licensure for Fewer Than All Conditions of Use for Which the Reference Product Has Been Licensed” outlining the procedure for such an application as well as adding a condition of use. Comments are open through April 6, 2020. On February 6, FDA issued a draft guidance on Biosimilar Licensing and Labeling, Timing of Certain Supplement Reviews which describes how companies can prepare draft labeling for biosimilar products and sets the timeline for consideration to a 6-month review cycle. Most significantly for communicators who work in the promotion of medicines, on February 3 FDA released a draft guidance “Promotional Labeling and Advertising Considerations for Prescription Biological Reference and Biosimilar Products – Questions and Answers” outlining guideposts for communication, particularly around making comparisons between biosimilars and reference products. The draft provides a range of Q&A that presents multiple examples of promotional language that is both appropriate and considered inappropriate by the agency. To underscore the importance of the action, FDA announced the draft guidance in a release that also announced a joint effort with the Federal Trade Commission (FTC) and the holding of a public workshop to be held March 9 at the FDA campus.
The approval and marketing structures have been primed by FDA to facilitate approvals and uptake of biosimilars. But there are certainly other impediments. For example, currently due to patent issues, several of the biosimilars that have been approved are not brought to market. A study published last year in the AMA Journal of Ethics (AMA J Ethics. 2019;21(8):E668-678. doi: 10.1001/amajethics.2019.668) of the 17 biosimilars approved, only 7 had made it to market. It is an example of only one of many issues that have consequences for uptake.
The more on the market, the merrier (for price). For generic drugs, the rule of thumb has been that the more that are on the market to treat a particular condition, the lower the price for treatment. But for biosimilars, we are not yet a point where there are enough on the market to have a significant impact on pricing. When it comes to biosimilar entries impacting price, we are looking at a process, not an event. FDA has worked to facilitate the process. The effects, however, would appear to be something for which we should be prepared to wait.
In the meantime, there are more near-term consequences for biosimilar marketing. The joint announcement by FDA and FTC not only sets up the guardrails for communications in the industry, it also is a signal of intent. The agencies will be watching closely and likely looking for a circumstance by which enforcement can stress the existence of the guardrails even more keenly. Despite the fact that enforcement by FDA with regard to promotional communications has been at historic lows, they agency seems to be saying pay attention. So pay attention.
Photo by Amir-abbas Abdolali on Unsplash
Share this:
January 23, 2020
Sorting it Out – FDA AdComm Review for 2019
Has FDA been holding enough AdComms? There have been a large number of drugs approved in the past year, but there has not been a corresponding increase in the number of advisory committees staged by FDA. Late in 2019, I had a posting about the fact that it looked like FDA was headed to a year where it held fewer FDA Advisory Committees than it had in years past. However, it was premature. FDA did manage to squeak in a few more meetings in December and brought the total up, though it was still well below the number of meetings held during many of the years between 2012 and 2019.
More about the volume of meetings in a moment. First let’s look at those that were held – what they considered and what the outcomes were. How man meetings, how many about drug approvals and how often was FDA in agreement with committees – and more…
There were a total of 28 meetings for the 2019 calendar yearOf the 28 meetings, 23 were held to consider an approval of a medicine, seven of which were sNDAs or sBLAs while the balance were NDAs or BLAs – the other five meetings were to discuss specific issues of safety or policy issuesOf the 23 NDA/BLA/sNDA/sBLA meetings, committees recommended approval for 17 medicines, did not recommend approval of 5 and 1 of the meetings was cancelled;Of those 17 drugs that Advisory Committees approved during 2019, FDA approved 12 of them; however three of them that got committee approval were instances where FDA decided against the recommendation of the committee and did not approve the application; and there are two applications where a decision has not been announced;In fact, there were a total of 4 examples of FDA going against the recommendation of the committee – 3 where FDA declined to approve against a recommendation (mentioned above) and 1 where FDA approved the drug in spite of the committee’s recommendation against approval – that would mean FDA disagreed with the Advisory Committee recommendation just over 14 percent of the time Four of the meetings involved joint sessions with the Drug Safety Risk Management Committee, only one of which was to consider an NDA for a new productThe committee that met the most was the same as in 2018 – the Oncologic Drugs Advisory Committee met six times this year, though that was down from nine last year; the next highest level of activity was also the same as last year – the Antimicrobial Drugs Advisory Committee met five times (three of them product-related) down from six meetings last year.
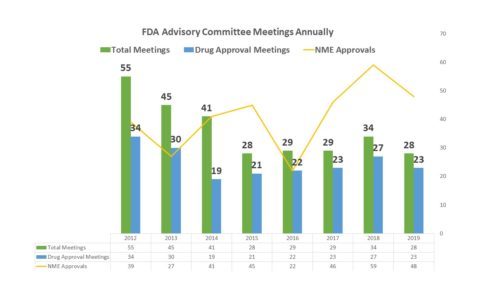
In addition to having a few more meetings at the end of the year, FDA approved a bunch of new molecular entities in the closing weeks of 2019 and in fact, is approving drugs more quickly, which was an aim of the policy changes made by Congress. Also, lately there has been some question regarding the higher rate of new approvals versus the lower number of FDA advisory meetings held. There have also been some approval decisions made by the agency that have also attracted criticism and concern. It is possible that this kind of scrutiny on the process could result in the staging of more advisory committees in 2020. We’ll have to see.
Photo by Laurynas Mereckas on Unsplash
Share this:
January 15, 2020
An Overview of FDA OPDP Enforcement for 2019
It is time to look back at enforcement for the 2019 year. It is still possible that FDA could post another letter, as there is a lag time between the time a letter is sent and the time it is posted to the web site, but let’s hope the Office of Prescription Drug Promotion (OPDP) did not send one out as a holiday greeting.
Comparing this year to other recent years, the enforcement actions taken by OPDP was not as sluggish as it has been, but neither was it robust. Long gone are the kinds of years where the agency would issue well over 100 enforcement letters for each and every violation that appeared. Instead now, the agency appears to have areas of concentration where enforcement is focused.

During 2019, as of this writing FDA issued a total of 10 enforcement letters, addressing communications carried in 11 different communications vehicles. While interesting warning letters went out from FDA outside of OPDP – for products containing CBD, for example, our focus is solely on promotional communications. Here are the highlights from this year:
Communications Vehicles – Violations were just about evenly divided between digital communications vehicles and traditional ones. On the digital side, there were 5 violations – the most frequent communications cited involved statements made on a website (4), and one letter sent regarding an email. On the traditional side of communications, there were a total of 6 – almost all DTC videos (4)- one of a DTC video involving a patient testimonial – one of the riskiest areas in promotional communications to my thinking. (I categorize DTC video in the traditional category rather than digital). On the traditional side there was also a banner ad and a print DTC ad involved in enforcement. Treatment Area – No single treatment area dominated enforcement. In the broad category of CNS-related treatments, there were two violations, two in oncology, two in erectile dysfunction. In short, there was nothing to indicate that the agency is focused on a treatment area with enforcement. Companies – Small companies watch out. The trend continues for OPDP to aim its enforcement mostly at companies that are generally not considered household names in the industry. This year was almost no different with the notable exception of the inclusion of Alkermes (more on that below). It is unclear whether large established companies have gotten highly adept over the years at scrubbing their communications to the point that they are more compliant with regulations or if FDA is finding that smaller companies provide good examples of violations the agency is concerned about. Warning versus Untitled Breakdown – Untitled Letters – also referred to as Notice of Violation (NOV) letters are considered the less serious of the two categories of regulatory action letters sent by OPDP, while Warning Letters are reserved for those violations FDA regards as more serious. This year, three out of the ten letters issued were Warning Letters. That is on par with a look at the cumulative rate over the years – in my database going back to 2004 that profiles over 330 letters from OPDP, about one in three letters are Warning Letters. So the proportion did not change this year. So even though FDA seems to be reserving its regulatory actions for violations it deems most noteworthy, that does not appear to alter the traditional approach to Warning Letters. Violations and Focus – There were 21 violations cited in the 10 letters issued this year. As per usual, by far and away, the most common violation cited by FDA is one that you would think might be the easiest area for compliance – the inclusion of risk information. This involved nine violations – nearly every letter issued by the agency this year. The other violations were for making a superiority claim (3), unsubstantiated claim (3), unapproved use (1) and promotion of an unapproved drug (2). That last one is noteworthy. While promotion of an unapproved drug was numerically low, it is proportionally high. Historically promotion of an unapproved drug has been an rather uncommon violation, having occurred only 19 times by my count since 2004, but 14 of them have been since 2012 and 11 since 2015, indicating it is a high area of interest for the agency in recent years. So the bottom line – there are two things to watch for in communications – risk information and pre-approval communications – particularly if you are a small-ish company.
Finally, one thing that occurred this year that was highly unusual with respect to enforcement concerning the final letter of the year issued by OPDP on December 2. FDA accompanied the posting of the letter – a Warning Letter – to the website with the issuance of a press release about the letter. That is highly unusual and in fact, I do not recall an instance where the agency brought so much attention to an enforcement action since April 2009 when OPDP (then called DDMAC) issued its misguided set of 14 letters impacting 45 brands. It does no good to second-guess reasoning, but it bears noting that his particular letter involved a drug utilized in an area where all stakeholders, including FDA, have been under scrutiny – opioids. The communications about the medication was lacking important risk information, FDA found. While risk information is obviously an important area of focus in enforcement, it is perhaps even more so for those manufacturers who are operating in the area of pain relief.
Photo by Goh Rhy Yan on Unsplash
Share this:
January 13, 2020
The Growing Profile and Influence of ICER

The year began with a prediction from Senator Mitt Romney to pharmaceutical executives that “change is coming”. There have been high profile hearings on the topic in the Congress and legislation that moved in both the Senate and the House of Representatives. There have been initiatives by the Administration in the wake of a blueprint issued but which has mostly gone nowhere. And the FDA has generated record numbers of approvals in generic drugs and issued policy documents to address two means of importation from other countries. All in all, there has been more talk than action.
But what of the talk? What did the conversation reflect from the past year?
Perhaps one of the most striking answers to that question is related to the emergence of the Institute of Clinical and Economic Review – ICER – from perhaps what could be described as the background of the issue of pharmaceutical pricing to the forefront. A non-profit that has been around since 2006 with a goal of providing a model framework for evaluating medicines for their value versus their cost, ICER has been steadily growing its presence in the media around the issue for a while. But this year in particular, the organization is not only being mentioned more in media coverage on the topic of pharmaceutical pricing, it is having an impact in actual costs and in coverage decisions. In short, ICER had not only a quantitative expansion of presence, but a qualitative impact as well.
This did not happen by chance. Part of the reason for the increasing profile of ICER in the media is of course that pharmaceutical pricing was a premium topic last year. But it is more than that. ICER issues a press release at every juncture of its milestone-ridden process and sends out a weekly report that summarizes its activity as well as overviews media mentions for the organization. In short, the organization has carefully cultivated its media profile.
 ICER has seen steady growth in its media profile, but 2019 was a marked increase in mentions in major publications
ICER has seen steady growth in its media profile, but 2019 was a marked increase in mentions in major publicationsIn a white paper published this week, my colleague Adam Silverstein and I examined the public discourse respecting pharmaceutical pricing in 2019. We look at media coverage of the issue – both the drivers of coverage and the stakeholders involved, in particular the ascent of ICER. You can download the paper here.
Photo by Adam Nieścioruk on Unsplash
Share this:
January 7, 2020
What They Said – A Look at Press Releases Issued by FDA in 2019
 In 2019 FDA Continued A More Robust Pace of Engaging Media
In 2019 FDA Continued A More Robust Pace of Engaging MediaIt is time to look at the forest rather than the trees. It has been a regular custom to occasionally look back and see what FDA has been saying, and how frequently the agency has been saying it and what the focus has been. During the entire year, we look at individual press releases, but let’s look at the entire body of communications. What is the same, and what has changed?
Naturally we start with a look at the total volume. This year FDA issued 272 press releases, down just a bit from the previous year of 289, but well ahead of the yearly output prior to that. It bears noting that 2019 began in the midst of the longest government shutdown due to an impasse on federal appropriations and which lasted until January 25.
Given that the volume has remained about the same this year over last, what about the subject matter? What did FDA talk about this year and were there any changes compared to years gone by?
Alerts – There were 13 press releases regarding alerts to consumers/patients issued this year, compared to 12 the previous year. This year there were fewer for drugs (4) and more for devices (8). Approvals – Despite the fact that there were fewer new molecular entities approved during 2019 than in 2018, there were more press releases about approvals this year (92) over last (74). This year there were 64 press releases about drug approvals, 25 about devices and 2 in tobacco and 1 in gene therapy. Last year there were 47 drug approvals announced and 27 in the device category.Legal Actions – This category includes issuing warning letters, seizures, and consent decrees and announcements were on par with last year – 36 in 2019 compared to 34 in 2018. However, the focus was different. This year there were more announcements regarding drug products over last year (17 in 2019, 8 in 2018) and less involving food (only 1 this year compared to 5 last year). There were also more involving devices this year compared to last (5 in 2019, only 1 in 2018). Recalls – Were about the same – 3 this year compared to 4 last.Rules and Guidance – There was an uptick in announcements of rules or guidance documents from 6 last year to 10 this year.
Commissioner Statements. All of the above categories are impacted to a degree by the presence of Commissioner Statements issued by FDA. Communications output from FDA changed dramatically once Commissioner Scott Gottlieb was at the helm of the agency when the number of annual press releases in 2018 increased a whopping 74 percent over the previous year and more than doubling the rate of 2016. This was largely due to the advent of statements issued from the Commissioner’s office. In 2018, there was an aggressive communications effort mounted from the Office of the Commissioner which began issuing statements from that office in the form of press releases. These offered a platform for the Commissioner to present a more personal point of view directly to stakeholders. Just prior to his departure, and since then, those types of statements continue to come out from the agency, but come from the heads of various divisions. Think of them like a tweet, but without character limits. Many of these showcased announcements of new rules, guidances and policy, while some were there to present a point of view about the agency’s plans or progress in a specific area of concern. The subject matter of these releases would not be captured in the numbers above. In particular, these communications devices are used to explain in greater detail or to spotlight a new guidance or rule that is going into effect.
What we do see about Commissioner Statements is the biggest difference between last year and this is the volume. While Dr. Gottlieb was still Commissioner in 2018, FDA issued 126 Statements from the Commissioner – a number that dropped to 89 for this year, many of which were statements not from the Office of the Commissioner, but from individual division heads within FDA. This is likely the biggest change of all in FDA’s communications as perceived through the issuance of press releases.
Spanish or No? FDA issues some press releases in both English and Spanish. It is not entirely clear, at least to this observer, when that happens and when it doesn’t. It has been difficult to discern a pattern, but last year there were 39 releases in Spanish as well as English, or 13 percent, which compares 32 releases this year which is about 11 percent.
FDA in Brief. One other thing of note. In 2017 FDA began issuing notices called “FDA in Brief” which are mini-missives to announce recent agency activities, but not up to a press release. One would think media and FDA beat reporters likely follow these. It is a mechanism FDA is using with greater frequency. In 2017, there were about 7 issued each month; in 2018 it went up to an average of 8 issued each month and in 2019 it was an average of 9.5.
The output from FDA in 2020 will be determined by the presence of a new commissioner, the lack of a government shutdown, and perhaps by the election cycle. We’ll keep an eye out.
Photo by AbsolutVision on Unsplash
[image error]
Share this:
January 2, 2020
2020 Vision – Notable FDA Actions During 2019

Happy New Year! It is 2020 – the year of clear vision. Let’s look back for a bit as we plunge forward.
FDA is a giant agency where a lot happens. Narrowing down the actions of significance is therefore a very subjective experience. Even limiting the subject matter to those actions which affect medicine, as opposed to food, devices and cosmetics, there is still a very wide bandwidth of activity and impact. And everyone gets to have their point of view. Having been involved in writing this blog since 2006, here is mine. Some of these noted items are things FDA has done – great milestones and achievements – some involve things the agency has not done – and others may involve situations that are evolving around the agency.
Fewer NMEs, But Interesting Approvals – At 48, there were fewer New Molecular Entities (NME’s) approved in 2019 than in the previous year when there were a whopping 59, but it was still more than in 2017, 2016 and 2015, thanks to a spate of approvals in December. That said, there were a number of interesting developments within the greater span of approvals, including some NMEs. These included the approval of two new treatments for Sickle Cell Disease, the approval of the first Gene Therapy (and incidentally the most expensive drug to be on the market); the approval of the third cancer treatment based on the presence of a genetic characteristic rather than tumor type; the approval of two new anti-depressants in a category that has long been without advance; and in vaccines, the approval of the first preventive for dengue as well as one for Ebola and the first test to detect Zika virus antibodies.

Three Commissioners in One Year – FDA started the year with Dr. Scott Gottlieb at the helm who surprised everyone with a departure in the Spring. In turn, he was replaced by the solid choice of Dr. Ned Sharpless who worked as Acting Commissioner for most of the year. Another surprise, despite the support of many past FDA commissioners, Dr. Sharpless was not appointed FDA Commissioner. Rather Dr. Stephen Hahn was sworn in as the 24th Commissioner of FDA. How the advent of a new commissioner will impact policy is anyone’s guess. During the confirmation process there was some question as to the commitment he has on the FDA’s efforts vis a vis the vaping industry, but as we learned during the Gottlieb confirmation process, concerns raised before don’t always materialize as deficits after confirmation. Even with the advent of a new commissioner, expect the media shadow of Dr. Gottlieb to extend to future FDA activities and policy announcements.
FDA Moves on Importation – Apart from generics, this is the other area where the agency can contribute to the effort to bring down the price of prescription drugs. It was an historic first in many respects – the first time the agency facilitated the pathway for the importation of drugs by issuing a draft guidance document and a rule. The former sets up a regulatory pathway for states and other entities to import drugs from Canada and the latter devises a means by which manufacturers can import drugs from outside the U.S. in order to lower the price in the U.S. though it remains a mystery as to why any manufacturer would likely do such a thing. The other historic thing about the announcement from FDA was that it was the second time the agency politicized the policy announcement by linking it to the Administration – the first being in September. For thoughts on that see the previous posting.
CBD Enforcement – CBD is everywhere. Everywhere geographically, everywhere in the span of products – teas, salves, dummies, hand cream, vape pens – you name it, CBD is likely there leading many stakeholders to label it the “Wild West” to signify that it is without oversight. In the wake of the Farm Bill which made legal the growth of hemp, FDA issued a press release in an attempt to preserve the agency’s regulatory authority over its use as an ingredient in supplements, drugs or food. That was despite the fact that the market was already running amok with products containing CBD. After his departure from FDA, Dr. Gottlieb penned an op-ed in The Washington Post outlining what FDA needed to do as the use of CBD was “getting out of hand.” During the course of the year, FDA issued a spate of warning letters to multiple manufacturers of a range of products including food producers warning them against the use of CBD. For an overview of FDA activity on this front, see “ CBD and FDA – Where Are We? ” Since that posting, FDA has issued additional letters. Can FDA send enough such letters to actually chill the CBD-related product market? Certainly the agency is under a measure of scrutiny given the opioid and vaping epidemics that are also high profile.
Record Number of Generic Approvals – The agency approved a record number of new generic drugs – one of the few contributions that FDA can make to impact the price of pharmaceuticals. The agency approved 1171 new drugs during the fiscal year. This follows on the heels of two notable years of approvals in 2017 and 2018 Notable announcements included the approval of a generic version of Advair Diskus, valsartan, naloxone nasal spray to treat opioid overdose, Gilenya – an oral treatment for multiple sclerosis, and the approval of a generic Eliquis in the atrial fibrillation treatment space. In addition, FDA issued multiple press releases during the course of the year providing updates on its progress to enhance the approval of generic drugs. With the backlog clearing out, it will remain to be seen if the pace continues in the new year.

And for 2020? Look for a likely uptick in attention to gene therapies as experience with those approved becomes more apparent and those in the pipeline get closer to the market. Watch what happens on the e-cigarette front given the concerns on the topic raised at the time of the new commissioner’s confirmation and monitor how FDA responds to the most recent criticisms of how the agency has handled the opioid epidemic. Probably best not to look for any further guidance on social media and communications by pharma as most of the research directed by OPDP is looking to traditional communications vehicles. As patients and the public have moved into social and digital for media consumption, OPDP remains with its head stuck in the sands of broadcast DTC.
Photo by Andrea Davis on Unsplash
Share this:


