
Mark S. Senak's Blog, page 12
August 24, 2018
Weekly Roundup 8.24.18

This is it. We are in the home stretch. Summer, at least the one defined by the school year, is almost over. Even though it is many years for me that I have been ruled by that calendar, it will always be the one most real for me. So even though Summer officially ends much later in September, really, this is it. We get one more week of respite from traffic before the Labor Day weekend signals the return to regular routine. And even though it is a time that is less productive than other parts of the year, here are a few things that happened this week of note.
FDA Announces Effort to Devise a Framework for Opioid Guidelines – The Commissioner issued a statement this week announcing that the agency has awarded a contract to the National Academies of Sciences, Engineering, and Medicine (NASEM) to assess the evidence that would be needed to inform future clinical practice guidelines for opioid analgesic prescribing are sufficient and to identify the research necessary to generate needed evidence. The scope of the work includes the commencement of a consensus study to identify where evidence-based clinical practice guidelines would best inform prescribers as well as a gap analysis of existing guidelines by, among other things, conducting a series of meetings and public workshops to engage contributing stakeholders in the analysis.
First Drug Approval for Rare Disease of the Cornea – Oxervate (cenegermin) won approval from FDA this week for the treatment of neurotrophic keratitis, a condition affecting the cornea in less than five in 10,000 individuals. It is, however, a degenerative disease that has a huge impact on the patient resulting in the loss of corneal sensation and progressive damage that in turn can mean thinning of the cornea, ulceration and perforation. Until now, treatment was palliative and involved surgery. Oxervate, a first-in-class recombinant human nerve growth factors, is a topical eye drop and is the first topical biologic approved in ophthalmology. Oxervate was a Priority Review, with Orphan Drug Designation, Fast Track and Breakthrough Therapy Designation. The company release can be found here.
FDA Extends Expiration Date on EpiPen – In the face of reported shortages for EpiPen and coincident with back-to-school time, FDA announced this week that it was extending the expiration date of some lots of the product by a period of four months and has put up a web page where consumers can check the numbers of the batches for dates. While it brings some relief to the shortage, along with the recent approval of a generic demonstrates the types of actions FDA can take to impact the status quo with regard to both supply and price. It also leads one to wonder what are the criteria for deciding what dates can be extended and how much wiggle room there is around a stated expiration date.
Things to Keep an Eye on This Week
August 29 – Senate HELP Committee Hearing – FDA Oversight: Leveraging Cutting-Edge Science and Protecting Public Health (Commissioner Gottlieb is sole witness)
September 4 – Public Hearing – Facilitating Price Competition and Innovation in the Biological Products Marketplace
Regulatory Developments in Pharma/Biotech/Devices
FDA Adds Four Tropical Diseases to Priority Review Voucher Program to Encourage Development
FDA Issues Draft Guidance – Hematologic Malignancy and Oncologic Disease – Considerations for Use of Placebos and Blinding in Randomized Controlled Clinical Trials for Drug Product Development
FDA Issues Draft Guidance – Osteoarthritis – Structural Endpoint for the Development of Drugs, Devices and Biologics
Photo by David Tostado on Unsplash
Share this:







August 21, 2018
Mid-Year Check on the Pace of New Drug Approvals at FDA

At the mid-year point, I usually do a check-in to see how we are doing on new drug approvals and how it compares to years gone by. I am quite tardy checking in on that front, so let’s get it done.
To set a little context, last year (2017) with 46 approvals of new molecular entities (NMEs) at FDA, it was a record year. However, it followed a year (2016) that was a little on the slow side with only 22 which, in turn, had followed another record year in (2015) with 45 approvals. So one year up, one year down, one year up.
Everyone is always interested in the number of new drugs being approved at FDA, but this year may be of particular interest because it comes in the wake of a broad policy push to lower the regulatory burden and to improve the pathway to support innovation. That is manifest not only in the passage and implementation of the 21st Century Cures Act, but in the repeated statements by the new Commissioner aiming at “modernizing” the approval process without sacrificing the agency’s gold standard for safety. It will be some time before that can be fully assessed, but in the meantime, many mechanisms have been put into place – such as Breakthrough Therapy designation, to enhance the regulatory process. One of our first insights into the effectiveness of these efforts is with the rate of new approvals for NMEs.
So at the first half of the year, things are coming along and we do not appear to be having the kind of dip we saw in 2016. In fact, FDA is at a slightly lower level of approvals than the previous year, but still going at a good clip. As of June 30, 2017, FDA stood at 23 approvals of new molecular entities, comprising exactly half of what would be the total for the year – 46. As of June 30, 2018, the number stood at 20, only two less than were approved the entire year of 2016. 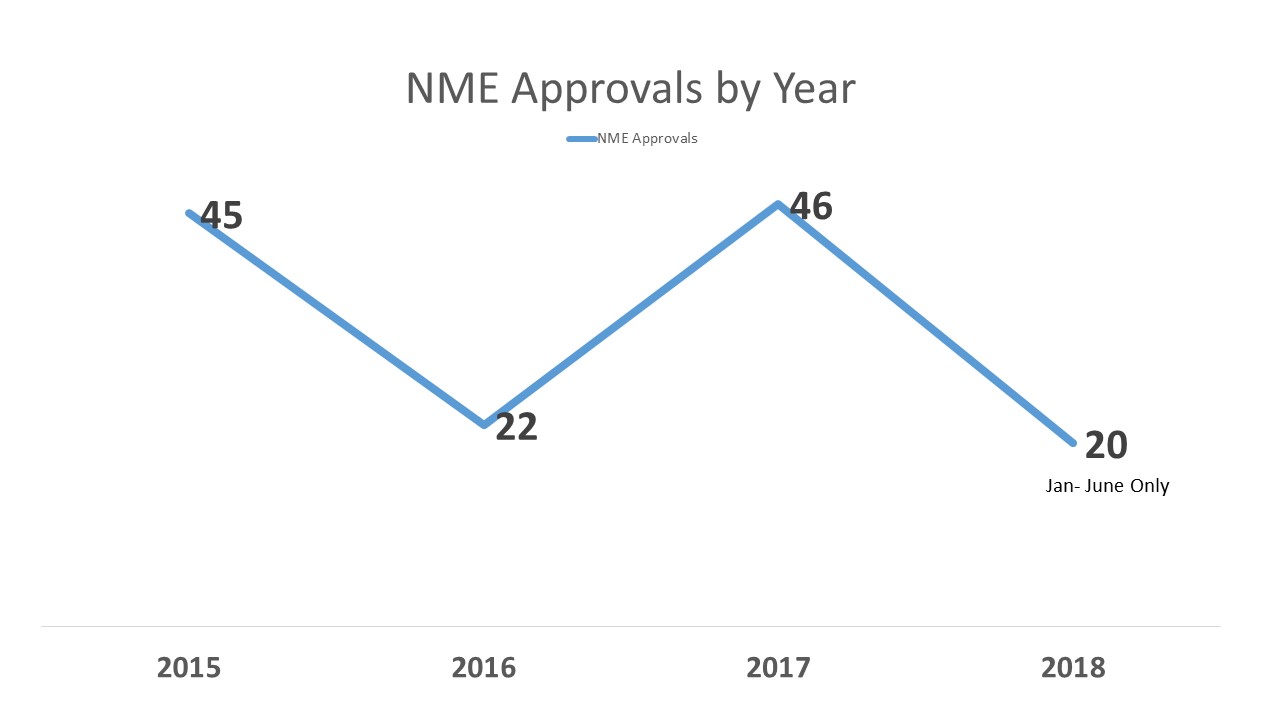
The number so far is high and if we keep the pace, the year should reflect a healthy amount of approvals. Will we do it? A few “surrogate endpoints” to look at to see how we are doing.
First, are there many advisory committees scheduled to hear drug NDAs and are they for NME? On that front, it does not look so hot right now – the advisory committee schedule page does not list any adcomms for drugs right now. Any that come much later in the year would likely represent PDUFA dates that will fall in 2019.
The other place to look is at known PDUFA dates. I run a little database on PDUFA dates gleaned from public sources such as company press releases. For the balance of the year, I have 42 PDUFA dates listed, but only 24 of them are for NDAs, as opposed to applications that are supplemental. That is not likely the entire universe of dates between now and the end of the year, only the ones that are apparent to me. Therefore the number is likely larger.
One final note given my lag in timing – since the mid-year mark, we have seen a healthy number of approvals. So in the end, we are likely to have a year that is on par, if not exceeding, last year’s number of NME approvals. Whether that is the effect of a healthy pipeline, the enhanced policy efforts in regulating the approval process or a combination of both still remains to be seen.
Share this:
August 17, 2018
Weekly Roundup 8.17.18

The vacation continues here in Washington, D.C. Traffic is lighter than a feather. Heat and thunderstorms continue and some of the leaves on trees have had it and are falling early, not in autumnal fashion, but as dried up little tired things – perhaps they were the earliest to appear in Spring and are now calling it a season. Tomatoes are still in their full glory which coincides nicely with bumper crops of basil. In the meantime, there was a little official life going on and here is some of it:
FDA Pronouncement on Opioids and Animal Care – Commissioner Gottlieb released a statement this week addressing the agency’s ongoing efforts to curb abuses in opioids but this time in relation to veterinary medicine. FDA produced a new resource to inform vets regarding potential abuse by humans with pets, designed to provide context around possible diversion by humans who are exploiting an animal in order to obtain opioids. The agency is recommending the use of alternatives to opioids where appropriate and advising vets on the development of plans should they encounter a situation where there is suspected diversion. The resource, however, refers vets to local police for information and does not provide a blueprint, guide or considerations for development of such a plan.
EpiPen Generic Approved – As headlines appeared regarding an Epipen shortage, FDA this week approved a generic version of the combination product auto-injector of epinephrine for use in the emergency treatment of allergic reactions. While FDA does not have authority over pricing of the products it regulates, the approval and introduction into the marketplace is one means by which FDA may influence pricing. According to the agency’s press release, this is the first approval of a generic version of the most widely prescribed epinephrine auto-injector in the US.. He added that the agency is committed to the development of generics, particularly of hard-to-copy products and would be advancing new guidance in this space as well as prioritizing review of generic applications. Last year FDA approved a record number of generic drug applications.
FDA, NIH on Modernizing Gene Therapy – In the wake of the approvals of the first three gene therapy treatments by FDA this year, the heads of FDA and NIH announced reforms to the review of gene therapy research designed to maintain safety while streamlining overnight, a regulatory reform theme during the past two years. Drs. Gottlieb and Collins stated that they would seek to reduce duplicative reporting requirements made by their agencies on research and that NIH would be dropping several of its review procedures though research will continue to be subject to FDA clinical trial regulations. To provide more thorough context and history regarding the gene therapy research and regulation, the pair published an article in the New England Journal of Medicine.
Things to Keep an Eye on This Week
August 23 – Senate HELP Committee Hearing – Prioritizing Cures – Science and Stewardship at the National Institutes of Health
No new FDA AdComms to report
Regulatory Developments in Pharma/Biotech/Devices
Voluntary Recall of 2 Thyroid Medications for Impurities
FDA Sets AdComm for Flu Vaccines Review for 2019
FDA Streamlines Procedure for Reporting Device Malfunctions
Photo by Annie Spratt on Unsplash
Share this:
August 10, 2018
Weekly Roundup 8.10.18

The summer creaks onward. In less than a month it will be Labor Day. I don’t know about you, but I find that almost incomprehensible. The folks on Capitol Hill are gone, which makes traffic here almost bearable. We like it that way. You can keep them all home if you like. One note related to the posting last week regarding the increase in the number of Commissioner Statements from FDA – this week there were none! That hasn’t happened since May. In spite of the quiet, some notable things have happened, so let’s saddle up and round up some news.
FDA Approves First DTC App for Prevention of Pregnancy – It is perhaps an historic first for Direct-to-Consumer as well as medical app support as well as contraception that FDA approved today a mobile app to support women in contraception to prevent pregnancy. The app works by providing support in the monitoring of conditions that would signal a likelihood of fertility on certain days – a method of contraception referred to in the FDA release as fertility awareness. Clinical studies to evaluate the efficacy of the app involved over 15,000 women who used the app for an average of eight months. Remember when you used your phone to make phone calls?
Approval in Non-Hodgkin Lymphoma – Non-Hodgkin lymphoma is a blood cancer that begins in the white blood cells. Two types of disease are Mycosis fungicides (MF) and Sezary syndrome (SS), both rare and difficult to treat. FDA announced approval of Poteligeo, a monoclonal antibody, with an indication to treat both of these types of non-Hodgkin lymphoma for patients with relapsed or refractory forms of MF/SS where there has been a prior systemic treatment . Poteligeo was given Priority Review and had Breakthrough Therapy status as well as Orphan Drug designation. The company press release on the approval can be found here.
New Generic Regulatory Pathway Sees First FDA Approval – FDA has very limited authority to impact the price of medications through its regulation of the pharmaceutical industry. However, the more generics that are on the market for specific conditions, then the more pricing pressure there is on manufacturers and generally the lower the price will go. With some treatment categories experiencing a lack of available generics, a new Competitive Generic Therapy Designation was put into place to expedite the development and review of a generic drug where there is a lack of competition. This week saw the first approval under that designation with the FDA approval of a generic potassium chloride oral solution for the treatment and prevention of hypokalemia (low potassium blood levels) in patients who are on diuretics. Ultimately pricing impact will depend on the number of drugs that utilize this category as well as the number of conditions affected. Time will tell.
FDA Expands Valsartan Recall – Back in July, FDA issued a press release announcing a recall involving some valsartan products prescribed for the treatment of high blood pressure. Since that time there have been a number of updates to the initial recall. The focus has been the presence of NDMA in some active pharmaceutical ingredient. The update from this week includes updated lists of products that are under recall as well as a list of products that are not under the recall. Both lists can be accessed in the most recent update. FDA is continuing its investigation and will provide further updates.
Things to Keep an Eye on This Week
No hearings
No AdComms
No FDA meetings
It’s August…..
Regulatory Developments in Pharma/Biotech/Devices
FDA Approves Drug for Prevention of Malaria
FDA Approves first of its kind targeted RNA-based treatment for rare disease
Photo by Jez Timms on Unsplash
Share this:
August 3, 2018
Weekly Roundup 8.3.18

This week’s posting about FDA Communications resulted in a nice story by Ed Silverman over at STAT and some Twitter action from the Commissioner about the role of the “Statement from FDA Commissioner” missives that I wrote about. Fun stuff. – On other fronts, as mentioned last week, I have been a little out of commission due to some personal things, but am back with my eye on things. Here is a bit of what I thought noteworthy from this week.
REMS Review – Dr. Gottlieb remarked via a statement on the meeting (held August 3) of the Anesthetic and Analgesic Drugs Advisory Committee and the Drug Safety and Risk Management Advisory Committee would be meeting to review the assessments of REMS programs, which were first approved in 2011, related to the use of transmucosal immediate release fentanyl (TIRF) products. The end goal is to determine whether or not adjustments to the elements of the program are necessary. The statement reaffirms the often expressed aim to strike a balance between the need for appropriate access versus potential for abuse. One focus of the meeting, he said, will be not only an examination of the data, but the accuracy of data, related to prescriptions of TIRF medications for patients who are non-opioid tolerant. For those interested not only in the issue of appropriate use of opioids for pain control, and the issue of misuse, but also those interested in how such data is accurately harvested, it should be an interesting meeting.
Warning in Women’s Health – Another statement from the Commissioner this week focused on a warning issued by the agency regarding the use of devices for “vaginal reconstruction” to treat conditions related to menopause, sexual function and urinary incontinence. The agency said that energy-based devices – commonly referred to as radiofrequency or laser – which have approval for specific conditions have been marketed for a non-approved use under the heading of vaginal reconstruction. The agency issued the warning for women and their doctors and notified seven manufacturers regarding the inappropriate marketing of devices.
House Energy and Commerce Letter to Opioid Manufacturers – This week the House Energy and Commerce Subcommittee on Oversight and Investigations sent a letter to three manufacturers of opioid products. The committee site excerpts from the letters states “For more than a year, the Committee has been investigating the potential breakdowns in the controlled substances supply chain which may have contributed to the nation’s opioid epidemic. Pharmaceutical manufacturers play a unique and critical role in this supply chain by researching and developing products for the consumer market as well as marketing such products after obtaining approval from the Food and Drug Administration (FDA)”. In short, the letters are seeking information, and pose a series of questions, aimed at gaining insight into marketing practices and awareness of misuse.
Things to Keep an Eye on This Week
Senate not in session.
August 7 – Antimicrobial Drugs Advisory Committee – meeting to consider NDA for amikacin liposome inhalation suspension for proposed indication of mycobacterial lung disease.
Regulatory Developments in Pharma/Biotech/Devices
FDA Approves iobenguane I 131 to treat unresectable, locallly advanced or metastatic pheochromocytoma or paraganglioma (rare adrenal tumors)
Photo this week by me!
Share this:
August 1, 2018
What They Said – FDA Press Releases 2d Quarter 2018
 A personal note – Apologies – I have been lagging on postings following a bereavement.
A personal note – Apologies – I have been lagging on postings following a bereavement.
As each quarter ends, it has been an Eye on FDA practice to look back and compare different types of activities to assess any changes or trends from FDA over the course of time. Today we’re going to look at the press releases for the first half of the year and at each quarter specifically, compared to the same time periods from last year.
If your own intuition has led you to think that the public is hearing more from FDA than they used to, then rest assured that you’d be right. The level of communication emanating from FDA is up dramatically over last year and over the first quarter. And the Commissioner is leading the way.
First of all, let’s look at the first half of the year. During the first six months of 2017, FDA issued 52 press releases, but for the same period this year, it more than doubled to 126 releases.
During the first quarter of 2017, FDA issued 23 releases and then saw a slight increase in the second quarter to 29. But in 2018, the number of releases issued the first quarter was 56 (more than double the same quarter the year before) and in the second quarter, that number increased to 70. That means that so far this year, not only has the overall number of releases increased, but so has the velocity.
What are they talking about – what leads to the increase? In the update on the first quarter of 2018, it was noted that the increase in the number of “Statements from the Commissioner” was the reason behind the spike. Such statements prior to the Gottlieb area were rare as hen’s teeth. Today they are exceedingly common and they are generally, though not always, employed to provide a depth beyond the issuance of a press release – to provide history and context around policy and to demonstrate progress – and to provide a personal stamp on agency developments by the Commissioner.
Here is a close up look comparing the first quarter of this year to last, and a comparison of the first 6 months of 2018 to the same period for 2017.
Approvals – You will note that while the total number approvals are slightly up for 2018 (32) over 2017 (30), drug approvals were actually down slightly (22 in first half of 2017 versus 19 in first half of 2018), while device approvals have gone up.
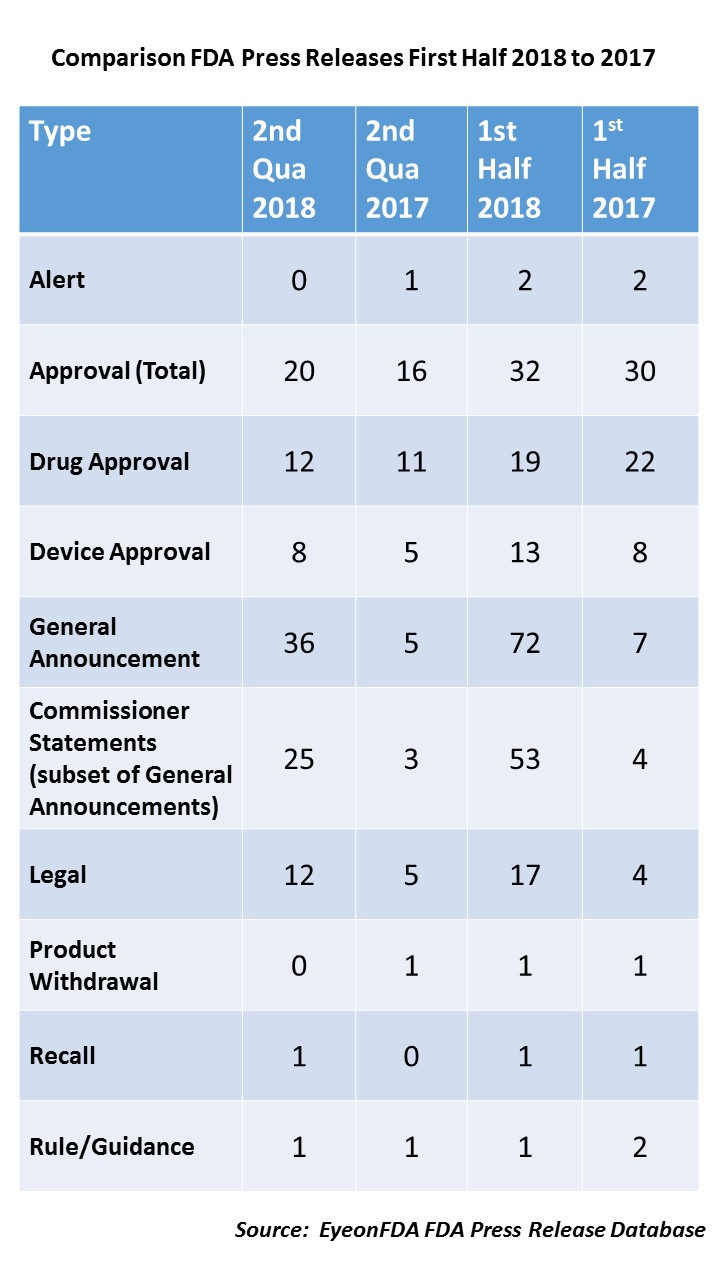
Legal Actions – Another big difference between last year and this was the increase in the number of announcements related to a legal action – a warning or consent decree – related in large part to actions the agency has been taking vis a vis kratom, e-cigarettes and opioid-related products.
Scott Speaks – Finally, the biggest change of all – Commissioner Statements. In the first half of 2017 there were 7 releases in the catch-all category of “General Announcements”, 4 of which were Commissioner Statements. In the first half of 2018, there were a whopping 72 General Announcements, 53 of which were Commissioner Statements, which is something like a 1200 percent increase. What does he talk about? Everything. He provides deeper background than exists in a press release about things the agency is doing – from the legal warnings on e-cigarettes to the reasoning behind new guidance documents and the policy implications of FDA actions, which may be announced in press releases, to provide context.
As noted in the report from last quarter, communications at FDA has definitely changed – there is more of it, and it is more in-depth. And the Commissioner is front and center as a driver.
Photo by G. Crescoli on Unsplash
Share this:
July 13, 2018
Weekly Roundup 7.13.18

Happy Friday the 13th, one of two for this year. Don’t worry, the other one happened already in April. I have been out on vacay for a bit, along with many I think. But back at the grind now and have some stuff coming up for you next week looking back at the first six months of FDA activity from a few perspectives. Congress is back from vacay as well, so things are on the upswing here in D.C., at least for a while. Here is a bit of what happened this week.
OPDP is on a Roll! Talk about vacations – after a long dormancy, this week FDA’s Office of Prescription Drug Promotion (OPDP) issued its third regulatory action letter this year, and the second for June. Like many others in the recent past, this one was for promotion of an unapproved drug. That means that 8 of the last 18 regulatory action letters issued by OPDP were issued about this violation, indicating that it is an area that will still move OPDP to act. The communications vehicles in question were an exhibit booth and a website. The compound is being studied for use in treating AML, but FDA said that the communications involved statements and presentations that made conclusions about safety and efficacy, using language to represent that the drug has a role in the treatment of AML. This letter was an untitled letter, as were the other two for this year – so far no Warning Letters. But just a reminder from OPDP, low enforcement does not mean no enforcement.
Gene Therapy Gets Guidance – A lot of it, in fact. FDA issued three new draft guidance documents and in addition issued updates to three existing draft guidance documents. The new ones related to the development of therapies in condition-specific circumstances. There was one for product development for hemophilia, for retinal disorders, and one for rare disorders. And because the field has seen rapid advance, those with new updates include Chemistry, Manufacturing, and Control (CMC) for Human Gene Therapy Investigational NDAs, Testing of Retroviral Based Human Gene Therapy Products for Replication Competent Retrovirus During Product Manufacture and Patient Follow-up, and Long Term Follow-up After Administration of Human Gene Therapy Products. In the past twelve months, FDA has approved three new gene therapies. You can read the Commissioners lengthy statement on the guidance documents here, and you can review and submit comments to the docket on any of them here.
Balancing Access to Pain Meds with Addressing Abuse – The line is not entirely clear. FDA has taken some meaningful actions to address abuse potential and some that might be considered questionable. This week there was an effort to provide an update and some context when the Commissioner issued a statement regarding access for patients in need of pain control with the need to take steps to stem abuse. While mentioning that FDA was holding a meeting with patients experiencing chronic pain in one of the agency’s Patient Focused Drug Development meetings and looking ways to innovate the development of devices that may treat pain, little was said in this statement about actual efforts being taken now to assure access for legitimate patients while a good deal of it was focused on avenues to misuse. In the end, to this reader, there was a lack in balance in the statement itself, much less the approach being taken.
Things to Keep an Eye on This Week
July 17 – Senate HELP Committee Hearing – How to Reduce Health Care Costs: Eliminating Excess Health Care Spending and Improving Quality and Value for Patients
July 17 – House Energy and Commerce Oversights and Investigations Subcommittee Hearing – Examining State Efforts to Improve Transparency of Health Care Costs for Consumers
Regulatory Developments in Pharma/Biotech/Devices
FDA Forms Drug Shortages Task Force
FDA Investigates Canine Heart Disease Potentially Linked to Diet
FDA Updates Warnings for Antibiotics – Flouroquinolone
Photo by Andreas Dress on Unsplash
Share this:
June 29, 2018
Weekly Roundup 6.29.18

Apologies are in order for the interruption in the Weekly Roundup schedule and no postings the last few Fridays. It was partially inspired by the fact that it was a busy few weeks for me. Conversely it seemed not so much for FDA despite the issuance of a number of not-so-interesting statements so it seemed a good idea to forego a few Roundup postings. Things got more newsworthy this week though and hopefully makes up for it. Included are a few things from weeks gone by. Next week no posting for July 4, resuming the week thereafter.
OPDP Issues 2nd Letter of the Year! Just when you think FDA’s OPDP is on permanent vacation, they surprise you. The office of the FDA in charge of promotional enforcement which used to issue dozens and dozens of letters to companies each year has eeked out its second for this year. This one was not a warning, but an untitled letter, and the subject was a video for a product that has a boxed warning on its label. Yet FDA found the video contained absolutely no risk information. The video depicted an interaction between a physician and a patient whereby the patient expressed a speedy and breezy use of the product, which FDA also maintained was not an outcome that was typical for patients who might use the product. You can see the letter here.
FDA Issues Guidance Clarifying Economic Discussions with Payers – In my last posting I covered new FDA Guidance related to discussions about useful drug information not contained in the label, but nevertheless consistent with the label. A companion guidance also covers discussions with payers and formulary committees about economic information not contained in the label to support decision-making around reimbursement. The audiences for this information include both public and private sectors, formulary committees, drug information centers, technology assessment committees, pharmacy benefit managers, third party administrators and other multidisciplinary entities that make coverage decisions. For approved drugs the information imparted must pertain only to approved labeling. The guidance covers situations where manufacturers are providing healthcare economic information (HCEI) to providers about approved drug products, medical devices as well as unapproved products, and the guidance divides the discussion under those categories.
FDA Approves First Medicine Derived from Marijuana – You would have to live under a rock not to have heard this news, but it is historically significant enough for a number of reasons to mention here. First, the newly approved drug, Epidiolex, a purified form of cannabidiol (CBD) and one of more than 80 active chemicals found in marijuana. The new approval, which lacks the THC component for achieving a “high”, is indicated to treat seizures as a result of Dravet Syndrome and Lennox Gestaut Syndrome (LGS). Both conditions affect children. It is the first approval for a drug to treat Dravet. Both conditions can have a dramatic impact on quality of life for those affected and their families. But significance is also derived from the fact that the source material – marijuana – has long been advocated as a potential source of medicine. The study for potential benefits has long been hampered by legal constraints and by dogma but FDA has provided guidance for the clinical study of botanicals. The approval was under Fast Track, Priority Review and the drug has Orphan Drug Status. You can see the FDA press release here, the statement by the Commissioner on the importance of research in this area here and the company press release here.
More on Patient Input – For a long time now we have heard about FDA transitioning to be more patient-centric, including the development and launch of a Patient Engagement Advisory Committee as well as holding a series of Patient Focused Drug Development meetings covering more than 20 disease areas. And in 2016, patient experience counted in the approval of a drug contrary to the recommendation of an FDA Advisory Committee. This week the agency issued a draft guidance document, the first of four planned, to explain to both patients and drug developers the principles by which FDA will incorporate patient and caregiver experience into the regulatory process. This particular guidance – Patient-Focused Drug Development: Collecting Comprehensive and Representative Input – concentrates on providing methodologies for how to operationalize the collection and use of patient experience data by drug developers.
Things to Keep an Eye on This Week
July 11 – FDA Public Workshop – Development of Treatments for Localized Prostate Cancer
July 12 – Antimicrobial Drugs Products Advisory Committee – Meeting to consider NDA for prevention of malaria relapse
Regulatory Developments in Pharma/Biotech/Devices
Split decision by FDA on candidate for two indications – one approved, not not
FDA issues guidance document on development of Pre-Exposure Prophylaxis in HIV
Photo by zelle duda on Unsplash
Share this:
June 14, 2018
A (Slight) Liberalization of Communications with Docs from FDA
![]()
What Happened. This week FDA issued a new final guidance related to communications about medical products that, while representing a slight liberalization of regulation when it comes to discussing useful information about a drug and the cleared uses for it, but which may not be included in the label. That said, the guidance does not address the issue of off-label promotions – or discussions that involve aspects of the product for uses in which it has not been cleared. The utility of the new guidance is that it will allow firms to talk about things – such as post-marketing study outcomes – that will enhance the understanding of the on-label use of the product.
What it Says. To shed light on where the parameters are, the guidance document provides eleven Q&A format discussions to demonstrate the approach that FDA is taking. The first Q of the Q&A covers which products are addressed and the second provides the crux the regulatory approach in answering how FDA determines whether a communication is consistent with the FDA-required labeling. The agency said that it uses three factors to determine compliance and failure to meet any of them results in a communication that is outside of the FDA-required labeling. The first such factor involves the actual information contained in the communication – whether it varies the indication, the patient population, affects the limitations and directions for use and whether it alters the dosing or use regimen. If the answer to any of these is yes – then the communication is not going to be consistent with the label. The other two factors are a bit less objective. The second of the three factors is whether the communications increase the potential for harm and the third factor is whether the directions for use enable safe and effective use under conditions that may be described in the context of the communication in question.
The balance of the guidance provide specific examples to more sharply define the parameters of how the agency applies the assessment of a communication by the company that is about information not included in the label, but which is not involving an off-label use.
Implications and Insights for Pharma. In considering how the new guidance may impact the communications by pharma companies, it is important to take into account where violations have occurred in the past. It is notable that FDA has issued this guidance during a time when enforcement by the agency is at a standstill with only a single enforcement letter having been issued by the Office of Prescription Drug Promotion (OPDP) so far during 2018 and following a record low of 6 last year. In reviewing FDA enforcement since 2004, out of the 324 letters issued by OPDP (or under its predecessor name DDMAC – the Division of Drug, Marketing, Advertising and Communications) by my count only 18 letters involved a violation of promotion of an unapproved use. Of those, 7 involved oral statements, mostly by sales representatives, though one involved a company CEO in a media interview. Other violations almost exclusively involved communications vehicles aimed primarily at physicians, such as Dear Doctor letters, direct mail or sales aids. So while the new guidance pertains to all communications by companies to physicians, because the most common violation occurred in the contest of an oral statement, particular care might be taken by companies to train spokespeople with regard to the parameters of the new guidance, especially for sales people who visit doctor offices or who are speaking with people at medical meetings while staffing exhibit booths.
Share this:
June 7, 2018
The Back and Forth on Safety Versus Speed and FDA

This week the Commissioner issued a statement related to the proposed modernization of the FDA’s drug review office, also the subject of a blog post by FDA’s head of the Center for Drug Evaluation and Research Janet Woodcock. While focused on the drug review process, they are just the latest pronouncements from the agency to address move to speed up the approval processes for all things – devices, digital health, artificial intelligence interventions, not just drugs. And with the passage and enactment of the 21st Century Cures Act among other things, there has been policy momentum to change the status quo. The word “modernize” has become code for reducing regulatory process – or sometimes in political parlance “reducing regulatory burden” without compromising safety or the “gold standard of the agency”.
There are a lot of good reasons to speed things up and to maximize access to new treatments. As Dr. Woodcock noted in her blog posting, the environment is dynamic – things change – evolving technologies, the emergence of new kinds of therapies – and the way we evaluating them has to change with it.
The need for changes in the system of approvals was very apparent in the earliest years of the AIDS epidemic when there were no treatments available to patients other than those used to address the opportunistic infections that came about as a result of a compromised immune system – and even those treatments were often without much impact. The average length of time that it took for a drug approval was much longer than it is today. According to a table developed by the General Accounting Office, in 1987 the average length of time from submission to approval for a new drug was at its longest – 33 months. By 1992 it was 18 months. In short the need was extreme, the ability of the system to deliver was not.
Though the wheels of progress moved slowly, as a result of AIDS activism, eventually changes came about that resulted in formalized mechanisms that had been under consideration to expedite drug approvals. The result were several new mechanisms introduced over a period of years designed to facilitate approval as part of the FDA review process – Priority Review, Fast Track, and Accelerated Approval. And the most recent pathway for expediting approval came in 2012 with the Breakthrough Therapy Designation.
But speeding approvals were also sometimes associated with at least a perceived sacrifice in safety. In the early 2000s there emerged a public debate about the impact of these programs when several high profile drugs were associated with serious side effects, some of which were eventually withdrawn from the market. Several drugs that had been Fast Tracked were withdrawn either for lack of efficacy or safety reasons such as hepatoxicity and cardiovascular events.
We are now on the cusp of a good deal of change that is poised to pick up the pace broadly.
As we approach a new level of a systemic easing the regulatory burden – one that goes beyond what was accomplished in the past in that regard- often coupled with reassurances that FDA’s gold standard will not be compromised – we have to be aware that getting the pendulum in exactly the right spot is likely impossible. Most of the focus so far has been on the steps being taken to speed up and facilitate the process of approval. But in doing so, we have to also ensure that our methods for evaluating that whether the “gold standard” is being maintained are also modernized so that we keep up with monitoring the effects of so much change.
Photo by Ben Ostrowsky on Unsplash
Share this:


