
Mark S. Senak's Blog, page 10
June 6, 2019
OPDP Takes Second Enforcement Action of the Year

This week FDA’s Office of Prescription Drug Promotion (OPDP) posted what was only the second enforcement letter of the year which was issued in May. Warning letters are issued by many parts of the FDA, but OPDP issues letters squarely aimed at the communication by pharmaceutical companies about the medicines they market. Twenty years ago such letters were issued with ferocious frequency, numbering over 150 at their peak. So far this year, there are two.
At least part of the reason for the drop-off has been a change in focus by the agency to be more risk-based in the expenditure of time, resources and effort – something former Commissioner Gottlieb felt strongly about. That has extended to OPDP which used to issue a letter for a technical infraction of communications boundaries that would not have really had an impact from a public health perspective, but are more prone to act now only where the agency sees infractions that are violations where there is a potential for some kind of harm.
That has meant, as noted here in past postings, that there has been a concentration on two types of violations. First, the presentation of risk information. And second, a focus on the promotion of unapproved products.
The most recent letter falls in the former category. There are two types of letters – Warning Letters reserved for situations FDA regards as more serious, and Untitled Letters for those considered less so. The letter issued this week was an Untitled Letter, sent to VIVUS for website content for the weight loss drug Qsymia. The letter was issued almost 15 years to the day that the company received their only other letter from OPDP (then DDMAC) back in 2004.
The letter regarding the Qsymia website addressed the presentation of benefit and risk information related to the product. Noting that obesity is an extremely common condition for which people are seeking help and that the drug itself had several limitations associated with its use and serious potential risks, the agency took issue with the balance between the benefit information presentation versus the placement of the risk information on the site. In addition, a claim that the drug worked three times faster than diet and exercise alone was cited by FDA as being a claim that was without support because the data cited for support calculated the amount of weight lost, but not the rate of the weight loss itself.
In addition to the risk information, OPDP addressed the treatment of benefits for the drug. The agency said that the patient data utilized in the discussion of benefits did not factor in the substantial number of patients who withdrew from participation in the clinical trials, which the agency deemed a selective presentation of results that resulted in overstating efficacy.
As stated above, this is only the second letter of the year. The prior one was directed at communication deemed to be pre-approval promotion. These are the two areas which seem to be the focus of the agency. While this letter dealt with obesity, the prior letter dealt in a product used in imaging related to treatment of oncology patients. Neither product had a boxed warning on the label, meaning FDA’s focus on areas where they consider a health implication to the violation is not confined to products with such labels. By OPDP standards, the year is still young and it is possible we could see more letters this year, but look for them in areas of risk information and promotion of an unapproved drug or use.
Photo by Goh Rhy Yan on Unsplash
Share this:







April 30, 2019
A Look at the FDA Revamped the Website

It has been a long time since FDA did much to its website. Being such a large and diverse agency, managing the site cannot be an easy thing. Aesthetics apart, just keeping track of pathways and names can be overwhelming and it is easy to lose track of what needs to be updated when things change. For example, one pathway to get to Warning Letters from the Office of Prescription Drug Promotion (OPDP) still listed that office by its former name from changed in 2011, the Division of Drug Marketing, Advertising and Communications (DDMAC). In addition, there were multiple pathways to get to some information. The information regarding advisory committees might seem straightforward initially but when one began navigating it, it was anything but.
But FDA has attempted more than just putting its (website) house in order, it also remodeled not only functionality, but the way it looks as well. The new site is much cleaner – the layout is far less busy and crowded and the look is much more contemporary. Just the other day I was wondering when they were going to get rid of that tired old picture of an advisory committee meeting and poof, it is gone.
So I am not a website expert, but I do work in communications and of course, am a frequent user of the site. Here are some takeaways:
User/Consumer Friendliness is Improved – FDA stated that one of the goals was to make the site more friendly to consumers and it certainly is an improvement. While the former site ran banner photos of some topical interest, the new site landing page features a topic with a large photo array that is focused on a feature topic. To get to the information, you need to click on the box announcing the feature, not the photos. Key to success for this approach will be topics that change out frequently as well as the topics themselves. This month’s is about children and allergy relief. Certainly topical given the time of year and the number of children, but perhaps not as serious as the recall of blood pressure medications. This approach may be more consumer friendly than the old site for sure, but the execution will tell just how much so. In fact, each division – Drugs, Food, Medical Devices has “featured information” that generally mirrors the FDA landing page (though stylistically there is not consistency across all divisions in terms of layout). For FDA to achieve its consumer friendly goal here they will have to work at providing information that is of interest to consumers and not necessarily just focus on that information FDA wants most to talk about. Longer, Less Crowded Landing Page – It used to be when you went to FDA’s landing page, you had a LOT of information crammed into the screen offering you pathways in a bunch of different directions at once – from links to speeches to advisory committee information to meetings information to the latest press releases, etc. All of that is still on the landing page, but it is more coherently laid out. That means that there is less splashed in your face on the screen, but the content has been elongated – and you now have to scroll down to find all the bits and pieces. That may not be entirely apparent to some. As you scroll down, you come to additional featured topics beyond the main one mentioned above. Right now one of them includes a link to information about the revamp of the site; a link to information about combatting opioids and one on FDA fostering drug competition. As noted above, these topics fall a little more into the category of things FDA may want to say versus the things we want to hear more about from a consumer perspective. As you scroll down, you come to press announcements (where curiously the title of the section is in smaller type than the titles of the most recent press releases). Scrolling down further takes you past many of the links that were formerly crammed into the landing square of the old site. It is almost all still there, just there more for your leisurely scrolling rather than in your face. But not everything is on the landing page. For that you need the next section.Menu Function is Key – In the upper right hand corner is the Menu Function. You are going to need this as it is the key to providing you a one-size fits all access to various divisions of the agency. It takes you to a site-map-lite that is actually very helpful if there are some specific things you want to look up. Most notably on the left side are a list of “featured links”. These are vital. They take you not only to guidance documents, but also one is the link that gets you to Advisory Committee information — to the page that is set up for each committee containing such information as the committee roster and meeting notices as well as documents related to specific meetings. To the right of this menu you will also find access to the FDA’s divisions, though it is not called that – instead it is called “Products”. Under that heading you will not see links to “CDER”, “CBER” or the others and where is the Office of Prescription Drug Promotion? Actually CDER, CBER and the others are there, but they are not called that. They are under their generic names (haha) – Drugs, Food, Medical Devices, Radiation-Emitting Products, etc. FDA may want to consider adding the acronyms here. These pages are generally laid out in similar fashion to the initial agency landing page, with a heading that is meant to cover topical information, prompting the user to scroll down to find the bits desired. Here you will find a link as you travel down the page to the Warning and Untitled Letters issued (still only one issued this year so far). The link to the Office of Prescription Drug Promotion exists but not on this page – it is under the About FDA Tab, and then drilling down through organizational structure through CDER there before you find it – here. Finding Specifics Related to Function in the About FDA Link – One thing you don’t see when you go to the Drug page or the Food page or any of the other division pages is a map for getting to where you want to go within that division (hence the lack of an OPDP link from the Drugs Page). To find that level of detail – to find a specific office that does a specific function, you may have to either conduct a Search (which can offer up a messy slew of links) or go to the About FDA link mentioned above. Here’s the thing – that is in very small letters at the very bottom of the landing page. It is a small, obscure link to an important function. They may want to think about elevating that one up to the top of the page, and making it a little more prominent.
All in all, as noted above, it looks a heck of a lot better and the navigation is less confusing now that everything is removed from a single frame shot. The order of things as you scroll down is pretty logical, though the demarcation of sections is subtle. It was certainly due for a change. Of note, if you have links to FDA materials at any site, some of the material may have shifted. On my own tab on Social Media and FDA, the guidance links appear to be in tact, but other links will need to be updated.
Photo by Jesus Kiteque on Unsplash
Share this:
March 13, 2019
Enforcement Updates: OPDP Issues First Letter of the Year
In recent years, FDA’s Office of Prescription Drug Promotion (OPDP) has been diminished in the volume of its enforcement expressed through the issuance of Warning and Untitled Letters. OPDP now sends out only a handful of letters each year when it used to send dozens and dozens (see below for year by year breakdown). In fact, for 4 out of the last 5 years, the number of letters has registered in the single digits. The reasons for this drop have been the subject of speculation in the past. Perhaps compliance is way up. Perhaps it is a political decision. Or perhaps – more likely – it is a focus of resources. Rather than ferret out each and every violation it can find, significant or not, FDA is maybe looking to issue letters where it can make a point or make a difference.
Recent enforcement trends, such as they are, bear that out. In February the office issued a letter recently posted to the FDA site directed to a sponsor and principal investigator in relation to an investigational new drug. The agency took issue with a webpage where the investigational compound was the subject of claims and presentations that the agency said made conclusory representations when the safety and efficacy had not yet been established.
The letter follows two patterns out of OPDP. A feature of enforcement letters over the past few years is that they are predominantly directed at entities that are not household names – smaller pharmaceutical companies or in this case an imaging center (though last year did include letters to Pfizer and Eisai). But the second, and more significant characteristic is the violation that is being more often cited – for the promotion of an investigative compound.
In a review of the 330 letters issued by OPDP since 2004, a letter issued for the promotion of an unapproved drub has been the subject of only 18 letters – 5.5 percent of all letters. Those 330 letters covered over 1100 violations (letters typically cite more than one violation) which means that promotion of an investigative compound has comprised only 1.6 percent of all violations.
However, if you look at the proportion that this particular violation occupies in the past few years, it tells a different story. Fully half of all of the letters ever issued by OPDP involving promotion of an investigational compound have come out since 2016, comprising one quarter of all of the letters issued 2016-2019, or nearly 15 percent of all violations. Both numerically and as a proportion, actions regarding promoting an unapproved drug has risen substantially, signaling that this is a current enforcement priority for FDA’s OPDP. If you drop 2016 from the mix, the result is even more pronounced with one-third of the letters being about an investigational compound and comprising nearly one-fourth of all violations.
Risk information – either presenting it in a way that minimizes risk or under circumstances that omit risk information entirely – still is the most common violation, but clearly promotion of an investigational compound has the focus of the agency just now. So bear in mind, low enforcement does not mean no enforcement.

Photo by Goh Rhy Yan on Unsplash
Share this:
March 6, 2019
Gottlieb Set a New Bar – What Happens Now?

The news that Dr. Gottlieb was leaving his post as Commissioner of FDA took everyone by surprise. Perhaps another surprise to many was the skill and judgment he brought to the job when initially he faced many critics due to his ties to the pharmaceutical industry. Yet of the many names floated at the time for the post, he was clearly the most qualified – offering the perspective not only of a doctor, but as someone who fought cancer, as a patient as well. And he brought experience. He was not new to FDA having served as a Deputy Commissioner under Mark McClellan. During that time, one would imagine that he devised a blueprint for how he would do the job were he Commissioner. When he actually became Commissioner, he went about implementing that blueprint.
But while detractors expressed concern over pharmaceutical company ties, as Commissioner it became apparent that what you saw with Dr. Gottlieb was what you got. Over the years as a Resident Fellow at the American Enterprise Institute he wrote extensively in national publications opining on a number of themes that were ones he would eventually bring to the job. In the grand picture, he looked at an agency that some may have said had become so entrenched in process that it lost sight of the goals for which it existed. The idea that everything should be done according to the same process it had always been done challenged progress. He embraced mechanisms that might loosen that up. Many of the policies that came about as FDA Commissioner he wrote about as themes in the years before.
For example, the idea of rolling submissions of BLAs and NDAs offered a means for companies to make progress in the development of new drugs at a faster pace. The tenure of Dr. Gottlieb as Commissioner is characterized by the notion that resources were best used in a way that maximized impact when it came to development and in the area of safety, focused on those areas where there was the most risk, rather than spread resources evenly across the risk spectrum. To that end, across a broad spectrum of development, there has been change effected in the way the agency is going about its business – from the faster development of generic drugs to new heights in the approval of new molecular entities.
Also during his time at FDA, the agency oversaw approvals of huge steps forward, particularly in oncology.
He has also not been shy to tackle large issues – vaping among youth and the opioid epidemic.
Moreover, he has not kept what he was thinking and doing a secret. Dr. Gottlieb is someone I’ve known for a long time and he always liked to talk. But as Commissioner, just as he did during his days at the American Enterprise Institute where he frequently published his opinions and ideas, he continued to do so through the issuance of “Statements from the Commissioner”. These were not one-offs. Rather there were topics, such as the development of a response to the opioid epidemic to the pursuit of more generic approvals to the development of pathways to speed the regulatory oversight over the approval process for new therapies, whereby he marked progress and communicated to stakeholders not only what was happening, but much of the reasoning behind it. Whereas past commissioners relied on a more reactive means of communications – providing updates during Congressional testimony and speeches for example, he took more of an initiative to be proactive. In doing so, he did it not only broadly speaking – on a range of topics, but with depth as well.
All of this changed what we have come to expect of someone heading FDA. The question becomes – now what? As he leaves the office, he demonstrated that one can have vision, ideas on how to execute that vision, and ability to do it when needed, all the while communicating transparently about it. He came to the office having had a long time to think about the role beforehand and he was effective in making change that had a positive impact. In addition, he is a snappy dresser. One hopes there is a similar presence that can keep the bar where it has been set.
Share this:
March 3, 2019
In the Pharma Pricing Debate, We Are Talking About More Than Apples and Oranges
 We are talking about more than apples and oranges….
We are talking about more than apples and oranges….This week the Senate Finance Committee held the second in what will be four hearings on the price of pharmaceutical drugs – Drug Pricing in America: A Prescription for Change. The first hearing, on January 29, listened to a panel of patients, patient advocates and academics, while the second invited the leadership of seven major pharmaceutical companies. In addition the House Oversight Committee on Government Reform held its first hearing of the year also on the topic on January 29, the same day as the first Senate Finance Hearing. Early in the year, Senator Mitt Romney reportedly told pharma execs behind closed doors that “change is coming” – similar words uttered by Senator Menendez at the close of the second Senate Finance Committee hearing. More talk is to come.
But in viewing both Senate Finance hearings, to the ear of someone who pays attention to communications, it becomes clear that at least part of the problem has nothing to do with rebates, or couponing, or any of the mechanics of pricing – it has to do with communications – what it is people are talking about. It boils down to three things – there is price. Then there is value. And finally there is cost. Stakeholders are speaking from different corners. They are oil, water, and something in-between. And they don’t mix very well.
Price. Patients are concerned about price. Patients, after all, are consumers before they were patients. As a consumer, you walk into a store and you see an item and you assess whether or not you want to buy it based on the price. If there is a comparable item at a lower price, you are likely to opt for that. If it is on sale, that is also a big factor. You expect that the store owner and the manufacturer are going to get a cut, and earn a profit, and you accept that as fair.
But when you are a patient, it is different. In the end, the patient perceives the medicine as it would any other commodity – like buying a new smart phone. He is willing to pay a premium price for the newest thing, appreciates that investment goes into developing the technology and expects that expense is written into the expensive, but ultimately affordable (for most) price. And above all, no one expects that they will have to ration food or paying utilities in order to buy a smart phone, the way that some patients testified in the Congressional hearings that they must do for medicine.
Value. Then there are stakeholders, like industry as well as organizations that assess price who consider the value that a treatment brings to the patient and to society as a whole. An example used during the second Senate Finance Committee hearing was in the treatment of Hepatitis C. Prior, many patients with this condition had to eventually undergo liver transplants – an intervention costly in many ways, but especially dollars. Therefore, a new medicine that brings a cure and thereby circumventing thousands of dollars in costs to everyone should be priced at a level that reflects that value. So if we are saving $50,000, a $25,000 price tag reflects the value that the treatment brings to the table.
The problem of course is that when there is a high price tag, many patients will have a hard time – high co-pays, high deductibles, mean high out-o-pocket costs. If it is a treatment for a chronic condition, this means high on-going costs. And the message to patients, who are thinking price – is that everything is being valued except their own well-being. It becomes a “your money or your life” proposition.
Cost. And here is where it gets a little murky. To address the situation, there are a whole set of mechanisms to adjust the amount that will ultimately be paid by the patients. There is the list price, the direct price (DIRP), average wholesale price (AWP), there are rebates, there is the wholesale acquisition cost (WAC). You practically – well not practically – you do need a guide for this part. There are enough acronyms to have the person of above average intelligence (PAAI) confused, frustrated and feeling like something is going on. And this is the part where pharmacy benefit managers (PBMs) come into play. For patients, these are faceless entities. They are also likely the subject of a future Senate Finance Committee hearing on pharmaceutical pricing.
When talking about this issue, each stakeholder is speaking a different language. Smartphone patient price perception is confronted by real estate value model and complicating it all are an entity we never see with a lot of confusing language.
In the end, what we have here is communications problem on top of the problem in our approach to this marketplace. This is one of those classic communications situations where many of the stakeholders are so focused on what they need or want to say, they are not taking into account what their audience needs to hear. One the one hand, if we are going to solve the problem, we have to make sure everyone not only understands the problem. But perhaps more importantly, if we are going to make real headway, it is not so much about government hearings spouting hyperbole, but rather taking into account who our audience is and what they value – determining what they need to hear and addressing it in the course of the conversation – that will ultimately bring about a more satisfying outcome.
Photo by Kawin Harasai on Unsplash
Share this:
February 27, 2019
FDA’s Contribution to the Pricing Issue – Generic Activism

As hearings on Capitol Hill these past two months (and in the months to come) have examined the issue of pharmaceutical pricing to inform potential legislation as well as to garner attention, FDA has been addressing the issue in the only way the agency can, through policies that enhance the development of generic and biosimilar competition for brand name drugs. In fact, in 2017, FDA approved a record number of generics – 1000 – a record the agency broke in 2018. Overall, the effort at promoting the development of generics as a means of addressing price has not only been assiduous, but wide in scope.
For starters, on Friday, February 22, the agency announced that it was releasing 74 product-specific guidance documents, 49 covering complex drugs, including 16 for which there are no approved generics with the aim of supporting approaches to developing new generics. It is one of many examples of how FDA has sought to move the needle in the generics market place. This is at least in accordance with a statement from the commissioner made after a GAO report released in early 2018 recommended that FDA revise its guidance documents on complex drugs.
Much of the activity is related to the fact that in May of 2017, FDA announced its Drug Competition Action Plan. Almost immediately thereafter (in June) published a list of off-patent, off-exclusivity brand name drugs that did not have competition as well as implementing a policy to help expedite review of generic applications. In that same announcement the Commissioner stated that FDA would expedite the application for a generic drug until there were three generics available for a brand product. In July of that same year, the agency held a hearing to identify where there were ways for the agency to enhance generic development.
In between the announcement of the Drug Competition Action Plan and the product-specific guidances, there was a good deal of other action announced or taken by the agency:
Policy Change – On the occasion of the release of the Trump Administration’s blueprint for addressing drug pricing, FDA issued a Statement from the Commissioner in which he reiterated the agency’s commitment to making an impact in the process for approving generics and in calling out “gaming” practices that might delay competitive generics in market entry and a few days later announced that the agency would be publishing a list of companies. In January of this year, the Commissioner issued a statement to signal the coming issuance of new policies to support generic entry into the market for complex drugs; Funding to Facilitate Generics – On February 13 of last year, FDA sought additional funding to, among other things, modernize generic drug review from text-based to data-based assessment. And in July announced the formation of a work group to examine a policy framework to look at importation of drugs address situations where there is only one U.S. approved and marketed version of an older drug that may not be commonly used, but is medically important and often involving generic medicines. Gaming Practices – Last Spring, the Commissioner announced steps on one of the issues touched upon in this week’s Senate Finance Committee Hearing on drug pricing – the “gaming” of the system to delay generic competition. The agency began publishing a list of companies identified as potentially blocking access to samples of branded products. That same month – also in a Statement from the Commissioner – FDA announced new policies to reduce the ability of brand drug makers to use REMS programs as a means to blog generic market entry. In October the agency announced new options to further curb “gaming” of the process by revising a draft guidance regarding the use of citizen petitions to add a resource burden to generic review process. Expedited Approval Pathways – In August the agency announced the first generic approval under the Competitive Generic Therapy designation – a new approval pathway created to expedite the development of new entries into the market where there is a lack of competition, formalized in an announcement this month and defined as a pathway granted to a company submitting an application where there is not more than one approved drug in the Orange Book. In October of last year in a Statement from the Commissioner new guidance documents were issued to advance the development of generic transdermal and topical delivery systems with the aim of enhancing the development of generic versions of complex drugs. Safety – This month, in the wake of media reports regarding the questionable quality of some imported generics, the Commissioner and the head of CDER announced steps regarding the oversight of safety and quality issues related to both domestic and imported products
As an agency, FDA has a narrow band-width in which to operate when it comes to the drug pricing issue. But within the space it has, the agency has been very active and very comprehensive. In fact, if you look for the word “generic” in the headlines of agency press releases between 2013 and 2017, you will find almost nothing. If you look at it in the past few years there are at least a dozen. If not activist, the agency has certainly been active.
Photo by Jack Harner on Unsplash
Share this:
February 21, 2019
Sorting it Out – FDA AdComm Review for 2018

Looking back at FDA Advisory Committee meetings (AdComms) held during any given year requires a bit of patience given that some of the approval results don’t come in for a while after each meeting is held. And in fact, there are still some drugs that were considered by advisory committees during 2018 for which there is still no FDA decision. But we are going ahead with a look-back anyway.
FDA holds advisory committee meetings in order to get input from experts and from the public regarding the approval of new products and has a fleet of advisors serving on those committees. For Human Drugs there are 18 committees with varied subject matter jurisdiction. Not every new drug application goes through the advisory committee process, but for those that do, from a communications perspective, it is often where the branding of a medicine takes its first breath.
That is because when there is an advisory committee meeting held it does bring more transparency to the approval process because there is a publicly open scrutiny of the application. This, in turn, has communications ramifications. While the application milestones and clinical study outcomes have largely been public, the bulk of attention has been paid in trade and scientific journals. When there is an AdComm, there is a shift to mainstream media coverage. And while the filing of the new drug application and the acceptance of that application are important milestones, it is when FDA posts the briefs of the professional reviewers within FDA that result in headlines in mainstream media. And because reviewers are citing their perceptions of the strengths and weaknesses of the drug in question, the comments in those briefing documents generate the first real round of headlines about the medicine. The second wave comes as a result of the meeting discussion and vote. And as we know, the vote is a recommendation to FDA which the agency may or may not follow.
All that said, how did 2018 stack up in terms of Advisory Committee activity and how did it compare to the year before? Last year, FDA approved a record number of new molecular entities (59). In addition there were approvals of biosimilars and approvals of drugs that were not new molecular entities, as well as label expansions.
More Meetings. There were more meetings held last year than the year before. In all there were 34 meetings of advisory committees last year, but only 27 of them involved the consideration of New Drug Applications (NDAs) or supplemental New Drug Applications (sNDAs). That is slightly higher than last year when there were 29 meetings, 23 of which were considering NDAs for approval, two of which ended up not taking place.
Most New Treatments Considered. In 2017, the Oncologic Drugs Advisory Committee came in at a whopping 9 meetings to consider drugs, but in 2018 met only once. The committee with the most meetings in 2018 was the Analgesic Drugs Advisory Committee with 7 meetings, indicative of the activity around pain. Also in 2018, the Antimicrobial Drugs Advisory Committee met 6 times.
Voting Patterns/No Votes. In 2017, the Committee that had the highest number and proportion of negative outcomes was the Arthritis Drugs Advisory Committee, voting down 2 out of 3 NDAs, while in 2018, it was the Analgesic Drugs Advisory Committee that voted down 2 out of 7. In 2017, the Advisory Committees voted to recommend approval 71.4 percent of the time; in 2018 it was 66.6 percent.
Disagreement in Outcomes Between FDA and AdComm. As is noted in every press release about an AdComm vote, FDA may or may not follow the recommendation of the committee. That naturally raises the question – how often does FDA go against the advice of an advisory committee. Consulting my own database that tracks outcomes, I have found that FDA goes against the advice of the committee about 11 percent of the time. In 2017, there were 21 recommendations and FDA went counter to the recommendation of the committee in 3 of them (14 percent). In 2018 there were 27 votes and FDA appears to have gone against the recommendation 4 times (15 percent). There is still one FDA decision still pending where FDA has extended the PDUFA date for the investigative treatment for postpartum depression Zulresso(TM) from Sage Therapeutics. If FDA’s decision ran counter to the AdComm recommendation of approval, would change these numbers slightly.
For your reference here is a table comparing the activity of the past two years. The first column denotes the number of meetings/medicines considered by each committee, the second the number of approvals voted, the third the number of times FDA had a different outcome from the recommendation.
 Comparison of FDA AdComm Activity 2017-2018
Comparison of FDA AdComm Activity 2017-2018Share this:
January 9, 2019
What They Said – A Review of FDA Press Statements for 2018

Periodically it is a good idea to take the pulse of what FDA is saying. This is not by looking at each press release individually, though of course that is important, but rather to look at the aggregate of what is being said – and how does it compare to the past? Utilizing a database created to track and characterize the output by the agency, I did a look-back at the end of 2017. Time to do so again. There are some interesting results.
Undoubtedly, this has been a banner year for FDA in many respects. There were more approvals of new molecular entities by the agency than ever and more approvals of generics. Communications is no exception when it comes to volume. This past year, FDA issued a total of 289 releases, a healthy increase over the 164 of 2017 and the 122 of the year before that. In short, FDA has had more to say – a lot more.
More Approvals – Fewer Approval Announcements. Given that there were more new molecular entities approved than ever, you might thing that the increase in approvals led the increase in communications. It did not. In fact, the number of press releases about new approvals actually declined during 2018. There were 74 releases about new approvals in 2018 compared to 77 in 2018, with a smaller proportion of those (47) being about drugs or biologics compared to 2017 (52) – that despite the fact that more drugs were approved in 2018. So more approvals, fewer press releases on that front.
What was the driver of the huge increase? It was, after all, a 76 percent increase (by the way I had Wolfram Alpha do the math). The primary reason for the increase was the number of “Statements from the Commissioner”. Last year he sent out 36 statements. This year he increased it to 126.
What is he talking about? Everything it turns out. A good deal of his focus was the enunciation of policy change for the agency that included departures from the past and setting up new regulatory frameworks – from the use of animals in laboratory testing to modernizing the regulatory process for the development of targeted therapies to new steps in the implementation of FDA’s pre-cert program. And related, he sometimes announces the issuance of new guidance documents. and has focused on marking progress and milestones respecting some particular issues, notably various aspects of the opioid misuse epidemic and FDA steps taken to address it. But there were also clearly issues that rose to the top – those he cared about personally (opioids and e-cigarettes) and where he was perhaps demonstrating that the agency was taking some issues seriously and acting upon them – including
e-cigarettes about which there were 4 statements over a two-month period; food safety – 4 statements about the Romaine lettuce safety issue from this year in addition to several more on food safety procedures in general;medical device regulatory approaches and approvals about which there were 6 statements;kratom – 3 statements about various actions around kratom, which is related to those he made about opioids;opioids – 11 more statements involving some aspect addressing opioid misuse and abuse – from new steps FDA was taking to new treatment approaches and much more
Certainly the Commissioner Statements allow the luxury to provide more context, background and reasoning than does a normal press release, and it was clear that there were a number of issues where he felt adding that context was helpful to explain to key stakeholders, most certainly including policy makers, not only why the agency is doing things, but seeking to underscore the announcement to demonstrate progress.
What Did FDA Talk About? When the communication wasn’t in the form of a Commissioner’s Statement, there were some definite increases in activity by the agency in some categories. For example, there were a much larger number of alerts about public health matters with 12 this year (romaine lettuce, e.g.) than last year with just 3. And there were a lot more announcements related to legal actions – seizures, warnings and the like this year (32) compared to last (19).
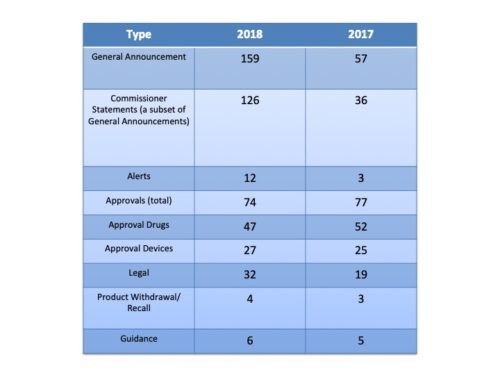 A Comparison of FDA Press Releases by Category 2017-2018
A Comparison of FDA Press Releases by Category 2017-2018Note that the Guidance category does not include at least 5 Commissioner Statements that were in regard to new guidance documents the agency was issuing.
Much has changed at the agency over the past year and a half. There has been a concerted effort at policy change. But not to be overlooked, and a point made in a prior blog posting, is that communications is among those changes. It certainly seems worth keeping an eye on it.
Photo by AbsolutVision on Unsplash
Share this:
January 2, 2019
Happy New Year – and FDA Statement on Government Shutdown

Happy New Year. On a personal note, I have been on a hiatus or shutdown myself to take time while undertaking a move. With that behind me, I am back at it and will be posting again with intermittent content and Weekly Roundups. And while I’m back, the government is not, having experienced a shut down just before the Christmas holiday. FDA has issued a statement on how the partial government shut down would be affecting the agency. In addition, the agency posted a Q&A for employees. The description of activities is not dissimilar to those outlined during the government shutdown of October 2013.
The statement provides an outline of activities that will continue, but there remains some vagueness about what functions will continue during what is referred to as the “lapse period”. The following are areas mentioned specifically:
Vital activities” to the extent permitted by the law, critical to ensuing public health and safety – however what exactly constitutes vital and what the law permits are not entirely clear.Maintaining core functions to handle and respond to emergencies – such as monitoring for and quickly responding to outbreaks related to food borne illness and the flu, supporting high-risk food and medical product recalls when products endanger consumers and patients – presumably this means that the core capabilities will continue to function, however, efforts such as recalls require coordinated communications efforts from the agency and it is not clear if this component is available from the statement. Pursuing civil investigations when we believe public health is imminently at risk and pursuing criminal investigations, screening the food and medical products that are imported and addressing other critical public health issues and surveillance for significant safety concerns with medical products continues – This sounds like it covers a lot of ground but turns somewhat on how terms like “critical public health issues” are interpretedSupport of activities funded by carryover user fee balances – It is somewhat uncertain as to what the term “support” means in this case. For example, advisory committee meetings to consider new drug applications would presumably be a user fee activity. An examination of the FDA advisory committee calendar during the shutdown of 2013 shows that meetings considering new products did take place. However, meetings that were not involving new product considerations were postponed. FDA has an advisory committee meeting on January 11 to consider an sNDA. See Dr. Gottlieb’s tweets noted below for more information in this regard.
Noted FDA activity and inactivity since the shutdown:
No new press releases issued, no FDA Voice Blog postings and remarkably, no Statements from the CommissionerFederal Register notices from FDA have continued to be published, though at a diminishing rate and publication might represent material already in the pipelineNo new warning letters issued out of any officeUpdates and clarification are being provided by some key personnel on Twitter. While some have only tweeted excerpts from the official statement, Dr. Gottlieb has been using Twitter to provide clarification on finer points on his Twitter feed.Some of Dr. Gottlieb’s pertinent tweets:From December 28 – We have questions re: drug review work that’s not user fee funded and won’t continue during shutdown. CBER will pause non-emergency work on whole blood, blood components for transfusion, allergenic extracts and HCT/Ps regulated solely under sec. 361 of the PHSA. For our Center for Drug Evaluation and Research this also includes pausing all OTC monograph drug activities. Only emergency work related to these products will continue.The 30-day review clock for any pending, non-emergency IND for a medical product that is not covered by a user fee program will be suspended during the lapse period. The clock will resume when the lapse period is over. However, new emergency INDs and IND amendments that relate tot he safety of individuals who are participating in clinical trials will continue to be reviewed during the government shutdown, even for products that are not covered by a user fee program. Many asked if FDA can accept new medical product applications during the shutdown. The FDA can’t collect FY 2019 user fee payments during the shutdown, which means we can’t accept new application for products under user fee programs: PDUFA, GDUFA, BsUFA, MDUFA, ADUFA, AGDUFA. FDA retains limited carryover balances for 21st Century Cures and opioids funding. These balances will only be spent on activities for which the funds are authorized. FDA carryover user-fee funds also allow the agency to continue work on existing user-fee related applications. (emphasis added by me)
Will endeavor to keep you posted on new developments. Happy New Year to all.
Photo by Craig Whitehead on Unsplash
Share this:
November 9, 2018
Weekly Roundup 11.9.18

So much to talk about, so little time. The election (thankfully) is over. The days are shorter, darker, wetter and altogether more lean. Thanksgiving is but a few short weeks away and Christmas decorations are already sneaking their way into stores and our psyche. There is a rush to finish all that needs to be finished before the end of the year, and perhaps it is no different at FDA where there has really been a good deal of activity over the calendar year, much of which we will visit in some year-end look-backs. In the meantime, here is what happened this week.
FDA and Expanded Access – Expanded access to drugs under clinical study is important on many levels – access for the patient, protecting the integrity of clinical trials, and of course patient safety. In a statement this week, FDA Commissioner Gottlieb announced improvements to the expanded access program that streamline the process even further by requiring less documentation and involving fewer individuals in the approval process. The agency also clarified how adverse events experienced through the expanded access pathway would be considered in the context of the review process for investigational treatments. In addition, changes have been made to improve the usability of the expanded access website, reducing duplication and adding new pages of commonly requested information. The statement also clarified that this program is distinct from the “Right to Try” legislation signed into law earlier this year and that FDA is in the process of considering what steps are needed to implement that legislation.
OTC Asthma Medicine Returns – Commissioner Gottlieb and CDER Director Janet Woodcock issued a joint statement this week about the return to market of a new version of Over-the-Counter Primatene Mist, the only OTC metered-dose inhaler for use in providing temporary relieve to mild, intermittent symptoms of asthma. It is approved only for persons who have been diagnosed with asthma by their healthcare provider. While there was a former version of OTC Primatene Mist, it was removed from the market in 2011 due to the fact that it contained chlorofluorocarbons (CFC) propellants known to be harmful to the earth’s ozone layer. In the wake of the removal, the two said that there were considerations as to whether having an OTC asthma product improved access for patients or actually caused patients to forestall needed treatment. They acknowledged that there is a narrow population of people diagnosed for whom an OTC asthma inhaler would have benefit, but that appropriate use needed to be reinforced.
Deadline Today on FDA Response to Senator Inquiries on Pre-Cert Program – On October 10, 2018, Senators Warren, Murray and Smith sent a letter to Commissioner Gottlieb and to CDRH Director Jeffrey Shuren containing a long-list of inquiries about the nature of the FDA pre-certification program for software being used in medical devices. The letter included nearly two dozen questions on a range of topics including particulars about the legal underpinnings of some of the aspects of the proposed pilot program that FDA is developing to allow specific manufacturers to facilitate the pathway to market for software used in some medical devices. The deadline for FDA’s response was today, November 9.
Things to Keep an Eye on This Week
November 13-15 – FDA Regulatory Education for Industry – Clinical Investigator Training Course
November 15 – Patient Engagement Advisory Committee Meeting – e-platforms
November 15 – Anesthetic and Analgesic Drugs Advisory Committee Meeting – opioid sparing outcomes in clinical trials of acute pain
Regulatory Developments in Pharma/Biotech/Devices/Food
FDA seizes food and medical products held in unsanitary conditions in Arkansas
FDA draft guidance on meta-analyses in clinical trials to demonstrate safety
FDA authorizes emergency use of fingerstick test for Ebola
Photo by Jarren Simmons on Unsplash
Share this:


