
Interesting new genetic study on insulin secretion and body mass
A new study uses a powerful genetic tool called Mendelian randomization to examine the relationship between meal-related insulin secretion and body mass. The results suggest that gene variants that increase post-meal insulin release may modestly increase body mass. Upon close examination, the findings do not appear to support the hypothesis that insulin is the primary driver of obesity, but they may nevertheless provide a foot in the door for insulin.
Earlier this week, I received a number of messages from people who asked for my opinion of this study. Here it is.
 Most people agree that bagels are fattening, but not everyone agrees on why.
Most people agree that bagels are fattening, but not everyone agrees on why.The study
The study was published in the journal Clinical Chemistry and was conducted primarily by researchers at Harvard medical school, including David Ludwig, MD, PhD, who is a middle author (1).* Ludwig has been a vocal proponent of the carbohydrate-insulin hypothesis of obesity, which posits that the primary cause of obesity is the ability of refined carbohydrate to raise insulin levels, trapping fat inside fat cells and leading to fat gain and excess hunger.
To understand the study, we first need to understand the technique it applies, called Mendelian randomization (MR). The concept behind MR is that nature randomly assigns gene variants within a population, similar to how a researcher would randomly assign people to treatment or placebo groups in a randomized controlled trial. This randomization process ensures that the variable of interest is not systematically associated with confounding factors, allowing us to say something about cause-effect relationships. If we know how specific gene variants impact some trait of interest, in theory we have a pretty clean experiment that tells us about how that trait impacts related outcomes.
Let me give you an example. The scientific community, including myself, believes LDL cholesterol level impacts heart attack risk. LDL cholesterol levels are partially genetic, and we have identified multiple gene variants that impact LDL cholesterol level. Using this information, researchers can see if gene variants that decrease LDL cholesterol are associated with lower cardiovascular risk (and vice versa). If they consistently are, this provides evidence that LDL cholesterol level contributes to cardiovascular disease risk. As it turns out, LDL-decreasing variants are associated with lower cardiovascular risk, supporting the hypothesis (2).
In the current paper, the researchers applied this technique to the relationship between post-meal insulin secretion and body mass index (BMI), a measure of weight for height that correlates with body fatness. Using large sets of human genetic data, they asked whether people who have gene variants that cause greater post-meal insulin secretion also tend to have a higher BMI. As their measure of post-meal insulin, they used insulin measurements taken from volunteers 30 minutes after they had consumed a glucose (sugar) drink.
The study did not have a preregistered research plan, meaning that we cannot be confident that researchers did not tweak the data analysis plan partway through to achieve a desired result, whether intentionally or unintentionally. To be clear, I’m not suggesting that the authors did so, only pointing out that they did not take the opportunity to prove that they didn’t engage in this common practice.
The results
The authors used three different groups of gene variants to estimate the impact of post-meal insulin release on BMI (insulin-30 stringent, GSIS stringent, and insulin-30 relaxed), and they tested them in two large genetic data sets (GIANT and UK Biobank). The key result is below:
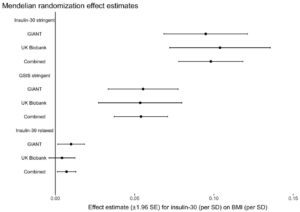
What this graph shows is that depending on the model used, one standard deviation higher post-meal insulin was associated with between about 0.01 and 0.1 standard deviation higher BMI, and this was statistically significant for most models. If I understand this correctly, roughly speaking it suggests that variation in post-meal insulin secretion explains between 1 and 10 percent of variation in BMI. About half of the larger estimate was driven by a single location of genetic variation, QPCTL/GIPR.
They then used the same method to test the reverse hypothesis– that BMI impacts meal-related insulin secretion– and did not find evidence for it, offering an argument that the previous association was not due to reverse causality.
Discussion
This study does provide evidence for a causal role of post-meal insulin release in BMI. However, before we get too carried away, we have to consider a few caveats and place the finding in the context of the rest of the scientific literature. Caveats:
The effect size is much smaller than carbohydrate-insulin proponents generally predict. Depending on the model, post-meal insulin explains between 1 and 10 percent of BMI differences, leaving 90-99 percent unexplained by insulin. This is not consistent with insulin being a major driver of obesity, although it does get insulin’s foot in the door.
The study measured BMI but did not directly measure body composition. We don’t know how much of the weight differences are fat, and how much are lean tissue. I would be less concerned about this if the effect size were large, because large differences in BMI are unlikely to be due to lean mass.
The regulation of glucose, insulin, and body fatness by the brain are closely related to one another (3). Since genes that are involved in one process are often involved in the other as well, it may not be possible to separate the two functions in a genetic analysis like this, meaning that the results could be (unintentionally) misleading. Although the authors tested for this possibility, they acknowledge that due to the inherent limitations of their data set, the test may not be very informative.**
Post-meal insulin secretion is associated with other physiological traits (possibly including leptin resistance***) that may also be influenced by the same gene variants in their models. It’s not clear that insulin itself is the variable impacting BMI, rather than another physiological variable associated with it.
When considered in the context of the rest of the literature, the current study’s finding is somewhat puzzling. For example, another recent MR study suggested that higher fasting insulin does not increase BMI, but higher BMI does increase fasting insulin (4). This result is basically the opposite of what the current study reported, but with fasting insulin swapped in for post-meal insulin. The authors of the current study suggest that the results differ because fasting insulin is simply a marker of insulin resistance, whereas post-meal insulin is a marker of insulin signaling exposure. This explanation doesn’t make much sense, for the following reasons:
Both fasting and post-meal insulin are strong markers of insulin resistance. If a person is insulin resistant, he will generally have higher fasting and post-meal insulin levels. As a matter of fact, the insulin response to a glucose drink is commonly used as part of a formula to estimate insulin resistance (5).
Fasting and post-meal insulin are correlated and respond to diet in concert. The amount of carbohydrate in the diet impacts both fasting and post-meal insulin. When calorie intake is matched, low-carbohydrate diets lead to lower fasting insulin in addition to lower meal-related insulin release (6).
So we are left with the puzzling fact that in the current MR study, insulin seemed to impact BMI, while in the previous study, it didn’t. I don’t know how to reconcile these two findings, and the authors’ explanation doesn’t do it for me.
Another puzzling thing about this study is the finding that BMI does not impact post-meal insulin release. The reason this is puzzling is that previous evidence shows unequivocally that it does! Check out this graph of post-glucose insulin before and after 4.5 months of moderate overfeeding (7). The closed circles are before, and the open circles are after:
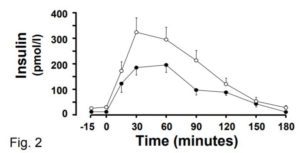
At the 30 minute time point (the one used in the current MR study), insulin secretion has nearly doubled, and this is after a modest weight increase of 2 BMI points. The fact that the current MR study did not observe this robust effect raises questions about how effective their method is at identifying cause-effect relationships in this domain.
Finally, carbohydrate-insulin advocates are using the current paper’s findings to support their larger hypothesis about the causes of obesity. However, there are still very good reasons to believe this larger hypothesis is incorrect, at least in its most prominent incarnation:
Ludwig’s own controlled trial suggests that a low-glycemic diet causes weight loss that is virtually identical to a high-glycemic low-fat diet, despite large differences in insulin secretion (8). This is consistent with the null findings of the longest and best-controlled diet trial of glycemic index I’m aware of, conducted by one of Ludwig’s colleagues at Harvard (9).
A 2016 study continues to be devastating to the idea that inhibiting the release of fat from fat cells increases hunger and body fatness (10). In a randomized trial, researchers gave volunteers with obesity a drug that substantially reduced fat release from fat cells, or placebo, for six months. No differences in calorie intake, macronutrient intake, metabolic rate, body weight, or body composition emerged.
The total rate of release of fat from fat tissue in people with obesity is higher than in lean people, not lower (11). Neither insulin nor anything else is trapping fat inside the fat tissue of people with obesity, making it hard to understand how obesity could be caused by this mechanism.
Rodents release insulin when they eat carbohydrate like humans, and their fat cells respond to insulin in the same way as humans. Yet diets that are predominantly fat tend to be more fattening in rodents than diets that are predominantly refined carbohydrate or sugar (12, 13). In fact, sugar is not very fattening at all in rodents, even at high levels. In every way that is relevant to the carbohydrate-insulin hypothesis, rodents and humans are constructed similarly, so why do rodents not respond to diet in the manner predicted by the hypothesis even under tightly controlled conditions?
Liraglutide is a weight loss drug that mimics the hormone GLP-1. This drug was originally developed as a diabetes treatment because it stimulates insulin release, yet it was extended to obesity therapy because it substantially reduces body weight and fat (14, 15). It also improves metabolic health (16). The increase in insulin is probably not the reason why fat loss occurs, but it certainly doesn’t prevent it.
Based on these results and others, I still feel fairly confident that the carbohydrate-insulin hypothesis as formulated by Ludwig and other public advocates is not correct. But could some version of an insulin hypothesis be correct? Perhaps. The current study offers the most persuasive evidence I’ve seen so far that insulin does impact body fatness, even if only modestly. It is consistent with a model whereby multiple factors, including post-meal insulin release, determine body fatness. If the effect of insulin is small, this may explain why it’s hard to detect in other study designs such as controlled trials of low-glycemic diets. It may also imply that the effect is of little practical relevance.
Due to the caveats I discussed, I’m not ready to endorse this model yet. I’ll need to see the finding replicated independently before placing greater emphasis on it. But this study has put a foot in the door for a model of obesity that includes post-meal insulin secretion, and I’ll endorse it if future research continues to support it persuasively.
* I’m not certain what role Ludwig played in the study, but some of the language and citations in the introduction are characteristic of his writing.
** “The Egger test was not statistically significant for any of the analyses, although this test is not well-powered and may not be valid in the context of substantial heterogeneity and/or violations of the InSIDE assumption.”
*** Higher post-meal insulin release is a marker of insulin resistance, and insulin and leptin resistance tend to be associated (and likely caused by overlapping mechanisms). I would guess that people with higher post-meal insulin release are also more likely to be leptin resistant.
Tweet

Share on Tumblr



Stephan Guyenet's Blog

- Stephan Guyenet's profile
- 40 followers

