
Mark S. Senak's Blog, page 47
May 1, 2013
Fewer Letters, Fewer Warning Letters
It may seem that I write a lot about the letters that FDA puts out regarding industry promotion and communication, but that is exactly what the nature of this blog is about. Other things can be interesting, particularly policy developments, but for those who work with medical products and communication, this is the heart of the matter and we have to look for patterns to gain insights.
Since putting together the database of NOV and Warning letters issued by FDA’s Office of Prescription Drug Promotion (OPDP), I have been looking to assess a number of patterns. Sometimes the meaning is obvious, in other’s I am left to speculate. This is one of the latter.
Every regular reader of Eye on FDA will note that they have frequently seen this chart evolve. Here is a profile of the combined pattern of Warning Letters and Notice of Violation letters issued by OPDP since 1997 and one will readily observe that the number of such letters experienced a steep decline after the 1990s. So that is the first point – there are fewer letters than there used to be.
 Looking at the same data up close for recent past, here is a more detailed version looking at the period 2004-2012, which is the span of time covered by the Eye on FDA Warning/NOV letter database.
Looking at the same data up close for recent past, here is a more detailed version looking at the period 2004-2012, which is the span of time covered by the Eye on FDA Warning/NOV letter database.
 When you break it down between the two types of letters – the more serious Warning Letter (expressed in RED) versus the NOV letters (expressed in BLUE), you also see that the proportion of Warning Letters has gone down. So far, for 2013, there are only 4 letters.
When you break it down between the two types of letters – the more serious Warning Letter (expressed in RED) versus the NOV letters (expressed in BLUE), you also see that the proportion of Warning Letters has gone down. So far, for 2013, there are only 4 letters.
 So, bottom line, the trend for the number of letters issued has gone down and the trend for the more serious type of letter has also gone down. Cause? I’m not sure. But would welcome ideas and thoughts from anyone willing to comment.
So, bottom line, the trend for the number of letters issued has gone down and the trend for the more serious type of letter has also gone down. Cause? I’m not sure. But would welcome ideas and thoughts from anyone willing to comment.
Share this:







April 26, 2013
Weekly Roundup – 4-26-13

The spring clothes are headed to the closet, while the winter clothes are headed to their space for their seasonal slumber until next Fall. The days are sunny. The winter mess in the yard has been cleared away (mostly) and new grass seed laid down. Cicadas will shortly be visiting the mid-Atlantic states….
Meanwhile, here is a bit of what happened in our world this week:
FDA Launches Effort to Protect Against Counterfeit Malaria Meds – This week, FDA announced the launch of a partnership to protect against counterfeit anti-malarial medications involving the use of a handheld detection tool developed by FDA called CD-3. The public-private partnership is with the Skoll Global Threats Fund, the U.S. Pharmaceopeia, the National Institutes of Health, the Centers for Disease Control and Prevention, the President’s Malaria Initiative, and the U.S. Agency for International Development. FDA said that the tool can detect falsified product, which can contain reduced amounts of drug that can result in the emergence of resistant strains, thereby posing a public health threat. The agency is working on improving CD-3 with additional refinements.
Senate HELP Committee Seeks Input into Draft Compounding Legislation – In the wake of continuing high profile issues related to compounding, the Senate HELP Committee posted draft legislation this week, seeking input into the draft which is due to the Committee by May 3 at 6 PM (ET). Among other things, the draft legislation establishes a clear boundary between traditional compounders and compounding manufacturers; clarifies that compounded drugs are new drugs subject to FDA jurisdiction; defines FDA’s role in oversight of manufacturers and sets up a user fee type system to fund inspections; and defines roles between states and the federal regulatory authorities. A link to the draft legislation is included on the newly created Eye on FDA tab that compiles resources related to the developing issues around compounding located on the blog.
Dr. Hamburg Opens Up FDLI Conference – Cites Budget Situation - Once again, FDA Commissioner Margaret A. Hamburg opened up the FDLI Conference here in Washington this week with an address that provided a comprehensive look at current agency activities and priorities. She noted that the times are challenging, though the challenges traditionally faced by an agency with such a large mandate is “made all he more difficult as we seek to fulfill our mission with the added pressure of the ‘new austerity’.” She said that FDA stands to lose approximately 209 million dollars this fiscal year – with $126 million in budget authority and $83 million of that in user fees because although the fees will continue to be collected, they will remain deposited with the U.S. Treasury. She also provided overviews on the status of implementation of key legislation such as the Family Smoking Prevention and Tobacco Control Act of 2009, the Food Safety Modernization Act and FDASIA as well as discussing aspects of the compounding issue.
That’s it for me this week. Get those closets switched around everyone.
Share this:
April 25, 2013
Warning and NOV Summary – 1st Quarter 2013
Each quarter I regularly try to provide a little insight into OPDP’s current thinking as expressed through the issuance of Warning and NOV letters. What we saw last year is that the number of such letters after rising a bit in 2009 and 2010 in number, slumped again in 2012.
But that is nothing compared to the first quarter of 2013, where we saw a total of 3, count ‘em 3, letters issued from OPDP to industry. Despite the fact that this quarter provides slim pickings, we persevere.
From this quarter, the three letters involved only three communications vehicles, none of which were digital properties (two were brochures) and none of which were Warning Letters and none of which involved drugs with boxed warnings.
The category for most common violation is usually Risk Minimization and while that did get the most this quarter, it tied with Superiority Claims at 3 and 3. Also included in this quarter was one instance of Overstatement of Efficacy, one Unsubstantiated Claim and one Inadequate Communication of Indication, making a total of 9 violations for the quarter, or 3 per letter.
Will keep you posted on next quarter some time in July.
Share this:
April 19, 2013
Weekly Roundup – 4-19-13

It has been a really busy week with travel and work that last Friday I didn’t even get to the Weekly Roundup. The schedule has been so busy, leaving no spare time for either posting here on the blog or continuing to clear away the dead growth in the garden. We have had a few minutes of Spring that already seem tempted to ebb into humid muggy days that we should not get till Summer.
But here is a bit of what I saw that happened when I came up for air:
Compounding the Issues Around Compounding - Three developments transpired this week regarding the increased scrutiny around compounding pharmacies in the wake of an outbreak of meningitis due to sterility issues. Taking them in chronological order – on Monday FDA issued an alert about the lack of sterility regarding products from two compounding pharmacies and serving notice of an impending recall. Then on Tuesday, the House Energy and Commerce Committee held a hearing entitled “A Continuing Investigation into the Fungal Meningitis Outbreak and Whether it Could Have Been Prevented” that was held nearly simultaneously with the release of a report by the Committee entitled “FDA’s Oversight of NECC and Ameridose: A History of Missed Opportunities“. Dr. Hamburg’s written testimony is available on FDA’s Web site, as is her testimony from a previous hearing held by the committee on this topic in November 2012 as well as a hearing before the Senate. Given the lack of clarity about FDA’s authority with respect to compounding pharmacies, it is probably an issue that is going to continue to be the subject of further public discourse.
Eye on FDA Tab on Compounding – Accordingly I have developed a tab on the blog that will be a repository of links to key milestones in the compounding saga.
FDA Approves Abuse-Deterrent Labeling for Reformulated OxyContin, But No More Generics – The agency announced this week that it had approved updated labeling for reformulated OxyContin which has physical and chemical properties to help reduce abuse potential. Moreover, the agency said that since original OxyContin now would pose a higher risk, that the agency has recalculated the risk-benefit consideration and determined that the risks no longer outweigh the benefits and therefore will not be accepting any applications to make original OxyContin in generic form. In addition, FDA provided an updated information page entitled Opioid Medications that outlines agency steps and provides more detailed information about the class.
That’s it for me this week. I hope everyone has a good and safe weekend.
Share this:
April 11, 2013
Comparing Types of Violations Between Digital and Non-Digital
In the wake of publishing “FDA Communications Oversight in a Digital Era” in last week’s blog post, I have been asked some interesting questions. One of them sought to know whether there was a difference between the types of violations of non-digital (traditional) communications vehicles versus digital.
Again, the paper was based on a data base I compiled that tracks OPDP letters to pharma companies from the years 2004-2013, inclusive, covering nearly 1000 violations cited by FDA. For this study, I specifically examined the years 2008-2012 in order to capture the uptake of social media by both mainstream media as well as patients and industry. For the period, there were 173 letters covering 675 violations. Each violation was characterized as belonging to a non-digital communications vehicle (brochure, advertisement, spoken word, e.g.) or a digital property (website, banner ad, e.g.). There were 385 non-digital violations and 290 digital violations.
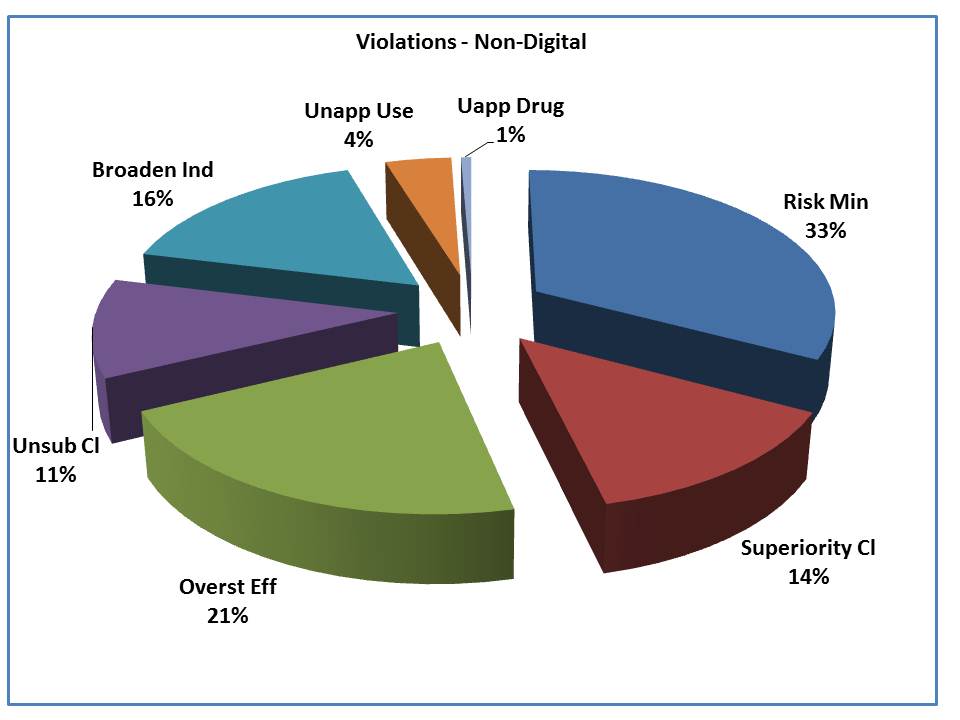
Here is a look at non-digital vehicles. You can see that 1 in 3 violations involve either the minimization of risk or the failure to include risk information. The second highest share of the violations goes to the overstatement of efficacy.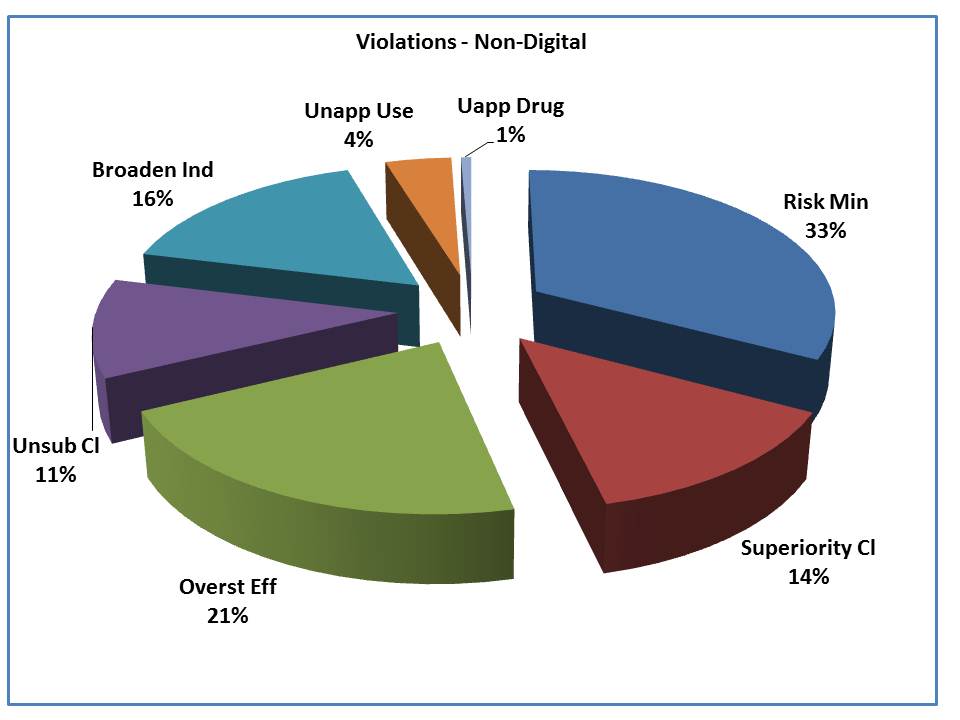
The breakdown shows a very different profile for the digital violations, where the failure to include risk information or minimization of risk had a much larger profile (56%) for digital properties rather than non-digital communications vehicles. Why?
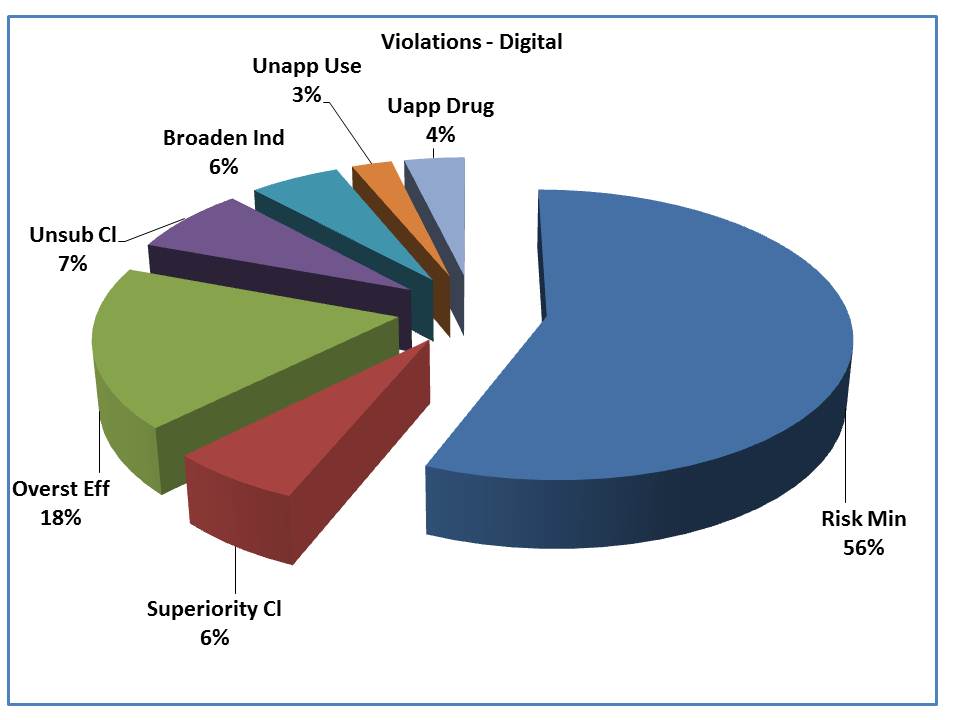 Remember 2009 when OPDP (then DDMAC) issued 14 letters regarding 45 brands for online ads? That added a huge number of risk-related violations since the reason those ads were being cited is that the “one-click rule” that everyone thought existed, in fact, did not. That artificially inflated the number of violations related to risk.
Remember 2009 when OPDP (then DDMAC) issued 14 letters regarding 45 brands for online ads? That added a huge number of risk-related violations since the reason those ads were being cited is that the “one-click rule” that everyone thought existed, in fact, did not. That artificially inflated the number of violations related to risk.
When you remove those violations, the two are more similar, though there remains a slightly higher level of risk violations as well as a higher rate of overstatement of efficacy and promotion of an unapproved product. With respect to the unapproved products, these involved 3 oncology products; one involved a Webcast and the rest were Website content.
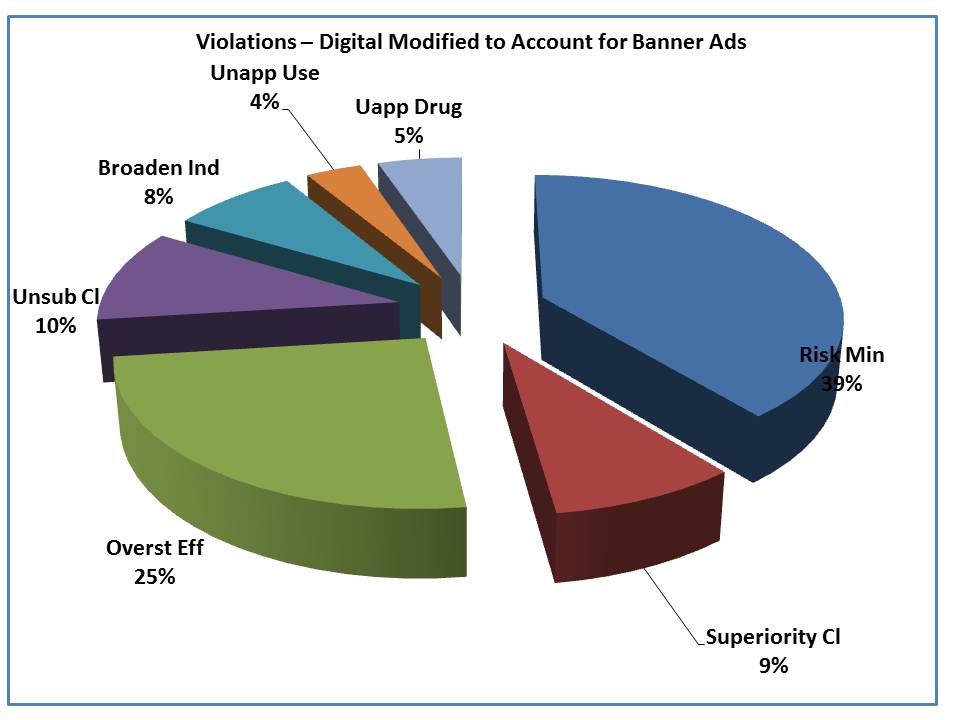 In the end, the old – it is the message, not the medium, seems to hold true – at least when comparing the types of violations between digital and non-digital properties.
In the end, the old – it is the message, not the medium, seems to hold true – at least when comparing the types of violations between digital and non-digital properties.
April 5, 2013
Weekly Roundup – 4-5-13

The daffodils are up and being rounded up in bunches and slipped into vases around the house, spreading good cheer, but overall Spring is still seemingly quite shy though indications are that is about to change. It is perhaps a good time to give the grill on the BBQ pit a good scraping for the coming season. It has been a mostly quiet week at FDA – perhaps everyone went away for a Spring Break! As of publication time, there were no press releases from the agency this week, now OPDP warning letters posted…. all quiet.
In the meantime, there were some interesting things:
Judge Rules Plan B Should Go OTC for All Ages – In a new chapter of a very long story of “morning after” contraception, a federal judge has reversed a reversal of a decision regarding the availability of an over-the-counter application for Plan B. In 2011, FDA was poised to approve the OTC application but that decision was historically ordered reversed by the intervention of HHS. Today that reversal was reversed by a federal judge and ordered FDA to approve the OTC application. Will this close the book on the subject?
H.R.1150 – Preservation of Antibiotics for Medical Treatment Act - The legislative developments are pretty much appearing here whenever I catch them. This bill was introduced in the House by Congresswoman Louise Slaughter (D-NY-25) with the purpose of addressing emerging antibiotic resistance as a result of use of medically important antibiotics to a food-producing animal for non-routine disease control.
FDA Commissioner Hamburg Speaks Out on Painkillers – In a blog posting on the FDA blog FDAVoice entitled “When Pain Relievers Cause Pain, Society Must Act“, Commissioner Margaret Hamburg addressed the abuse of opioids in the U.S. Stating that in “2010, an estimated 16,651 peopled (sic) died because of inappropriate use of prescription opioid drugs, a 313 percent increase over the past decade”, she outlined FDA’s approach which involved a blend of continuing research about pain, discovery of abuse-deterrent formulations, more accurate labeling, increased education, mandatory training programs for prescribers, new packaging and improving availability of new products that can treat abuse and overdose. On that same day, Senate Minority Leader Mitch McConnell sent a letter to FDA urging the agency to consider ways to ensure that tamper-resistant efforts taken by brand name pain killers are also in use by generics that come to market.
That’s it for me this week folks. I hope you all have wonderful weekends and see you next week, when surely Spring will have grown more emboldened.
April 2, 2013
Some Digital and Social Media Guidance – FDA Regulation of Pharma Communications in a Digital Era – A White Paper

I have been working to assemble a data base of FDA/OPDP Warning and NOV letters spanning the years 2004-present, tracking activity through a number of fields. The database now includes hundreds of letters and covers nearly 1000 violations cited by the agency with regard to communications by pharma about medical products.
I tracked several different fields of information. I can sort by company, time period, product, product indication, type of violation, type of communications vehicle, whether or not the product had a boxed warning, and for our purposes today, whether or not it was a digital communications vehicle (web site, social media e.g.) or a traditional medium (print ad, brochure).
 For some time, OPDP – and prior to that DDMAC – has been struggling to understand and respond to the regulatory challenges posed by new emerging digital communications platforms, with little to show for it. Accordingly there are a number of outstanding questions about how OPDP regulates communications on the Internet and digital platforms, including social media. For some in industry, the lack of guidance has had a chilling effect on participation in social media and even the Internet, despite the fact that it is a resource to which patients regularly turn for information.
For some time, OPDP – and prior to that DDMAC – has been struggling to understand and respond to the regulatory challenges posed by new emerging digital communications platforms, with little to show for it. Accordingly there are a number of outstanding questions about how OPDP regulates communications on the Internet and digital platforms, including social media. For some in industry, the lack of guidance has had a chilling effect on participation in social media and even the Internet, despite the fact that it is a resource to which patients regularly turn for information.
As a result, digital and social media have become a sort of regulatory bogeyman. Lacking any sort of formal guidance from the agency, the only peek into FDA’s point of view is to examine enforcement patterns. So I have used the data base to compare enforcement patterns vis a vis digital communications. For purposes of this paper, I narrowed the search to the years 2008-2012 (inclusive) to coincide with the ascendency of social and digital media use by pharma. For this period, I wanted to see how violations by digital communications properties compared to violations by traditional (non-digital) communications vehicles.
In short, I asked the data the following five questions:
What was the frequency enforcement among digital versus nondigital communications vehicles used by pharmaceutical companies?
What was the comparison of the number of violations involving nondigital communications versus digital communications?
Was there a greater frequency of more serious violations for digital versus nondigital communications vehicles?
In looking at a year-by-year breakdown, is the rate of violations related to digital communications increasing with the heightened use of digital and social media?
What has been the frequency of involvement of social media assets to generate regulatory action letters from OPDP?
You can download the paper here or by clicking on the photo of the cover above.
I hope that the answers to these questions provide a useful basis to help communicators in consideration of their own efforts to talk about medicines in today’s communications and regulatory environment.
The paper has its limitations. For example, the most obvious limitation – since we do not know what proportion of communications by industry is divided between digital and non-digital efforts, the data cannot say whether or not digital is over or under represented. In other words, if 50% of the communications effort is in digital, and it gets 50% of the regulatory action violations cited by FDA/OPDP, then it is proportional – however we have no way of knowing that context.
In the coming weeks, you will find additional analyses from the OPDP action letter data base here on Eye on FDA.
March 29, 2013
Weekly Roundup – 3-29-13

Spring has started, but not sprung. Not really. In the days since Spring first started a mere 9 days ago, we have had more snow in the Washington, D.C. area than we had all winter. Still, the daffodils have shown up, poking their heads through the white and looking actually somewhat energized by the bouts of snow and cold rain, which is more than I can say for most of us.
In spite of all that, here is a bit of what did happen in FDA-world this week:
FDA Approves New MS Treatment – The agency announced approval of Tecfidera (dimethyl fumarate) for the treatment of adults with relapsing forms of multiple sclerosis. For those suffering from MS, most experience episodes of worsening function initially followed by recover periods, but which may become incomplete over time which leads to a progressive decline in function.
An Alert Over Temporary Tattoos – After saying that it had received reports – though not saying how many – of serious and long-lasting reactions to the coloring used to provide temporary tattoos on the skin, FDA’s MedWatch issued a consumer alert about the practice – Temporary Tattoos May Put You at Risk. The agency said that the problems being reported included redness, blisters raised red weeping lesions, loss of pigmentation, increased sensitivity to sunlight, and even permanent scarring. The skin is the largest human organ – let’s take care of it!
Introduction of Safety and Fraud Enforcement for Seafood Act - It is H.R.1012 in the House and introduced by Congressman Markey and it is S.520 in the Senate introduced by Senator Mark Begich. Seeks to improve interagency cooperation on seafood safety and fraud protection. It seeks to define seafood fraud and requires data to be submitted regularly, particularly regarding frozen or farmed seafood; provides standards for refusal of admission of imported seafood and requires a public website to list exporters of seafood to the U.S. and to track violations. The Eye on FDA Proposed Legislation tab located at the top of the blog has been updated.
That’s it for me this week folks. I have a real good surprise for you on Tuesday so be sure you check in with Eye on FDA!
If you are observing Easter, have a good one. If not, have a lovely weekend. Let’s get on with Spring now….
March 28, 2013
Upcoming FDA Advisory Committee Meetings and Topics
In January Eye on FDA ran a look at the upcoming advisory committee meetings and their topics and it is time for an update.
It turns out that April is a ridiculously busy AdComm month, but there are some interesting meetings certainly.
![]()
April 5 – Orthopaedic and Rehabilitation Devices Panel of the Medical Devices Advisory Committee – the committee will be discussing the possible reclassification of Shortwave Diathermy devices, Class III, requiring premarket approval. Background information will be posted here.
April 8 – Ophthalmic Devices Panel of the Medical Devices Advisory Committee - the committee will discuss and make recommendations on information regarding the premarket approval application for the Trulign Toric posterior chamber intraocular lens sponsred by Bausch and Lomb. Background information will be posted here.
April 11 – Device Good Manufacturing Practice Advisory Committee - the committee will be meeting to discuss the potential effects of extreme weather and natural disasters on medical device manufacturing chain processes and marketed medical device safety and quality and how to optimize FDA’s current regulatory framework to address risk and vulnerabilities to manufacturing from extreme weather. Background material will be posted here.
April 17 – Pulmonary-Allergy Drugs Advisory Committee - will meet to consider NDA for fluticasone fuoate and vilanterol dry powder inhaler (proposed tradename BREO ELLIPTA) sponsored by GSK for the long-term maintenance of airflow obstruction and for reducing exacerbations in patients with COPD. Background materials will be posted here.
April 18 – Joint Meeting of the Advisory Committee for Reproductive Health Drugs and the Drug Safety and Risk Management Advisory Committee - to consider NDA for AVEED (testosterone undecanoate) intramuscular injection from Endo Pharmaceutical Solutions for the proposed indication of replacement therapy in adult males who have a deficiency or absence of testosterone. The safety discussion will focus on postmarketing reports of oil microembolism in the lungs and potential anaphylactic reactions. Background information will be posted here.
April 25-26 – Clinical Chemistry and Clinical Toxicology Devices Panel - meeting on the 25th to discuss and make recommendations on the appropriate regulatory classification for diagnostic devices known as methotrexate enjyme immunoassays which are considered pre-Amendment devices in commercial distribution prior to May 28, 1976. FDA will be seeking safety and effectiveness input. A second session being held that day will look for recommendations on the appropriate classification for diagnostic devices known as phencyclidine (PCP) enzyme immunoassays and PCP radioimmunoassays, also Pre-Amendment devices and FDA is seeking safety and efficacy input. On the 26th it will be the same drill for isoniazid test strips, also Pre-Amendment. Background material will be made available here.
May 2 – General and Plastic Surgery Devices Panel - the committee will discuss and make recommendations related to the premarket approval application for the Juv[eacute]derm Vouma XC sponsored by Allergan – a dermal filler indicated for deep implantation to restore lost volume. Background meeting information will be posted here.
May 2 – Oncologic Drugs Advisory Committee – Two NDAs are up for consideration. In the morning the committee will discuss AVEO Pharmaceuticals NDA for tivozanib capsules with the proposed indication for the treatment of advanced renal cell carcinoma. In the afternoon Delcath Systems NDA for a drug/device combination with the proposed trade name Melblex Kit which is melphalan for injection to be used with the Delcath Hepatic Delivery System for the proposed treatment of patients with unresectable ocular melanoma that is matastatic to the liver. Background materials will be posted here.
May 3 – Joint Meeting of the Medical Imaging Drugs Advisory Committee and the Oncologic Drugs Advisory Committee – meeting to discuss the safety and efficacy of currently approved leukocyte growth factors (LGFs) as potential treatments for radiation induced myelosupression associated with a radiological/nuclear incident. Background material will be posted here.
That’s it for now. I will update periodically.
March 22, 2013
Weekly Roundup! Back again! – 3/22/13

I miss doing the Weekly Roundup and so I am bringing it back. Part of the reason that it subsided is that I have been working hard on a rather large project that is now concluded, so let’s round up the news again once a week. Here goes.
Senator Franken Introduces the FAIR Generics Act – The Fair and Immediate Release of Generic Drugs Act was introduced in the Senate and would revise the definition of “first applicant” for purposes of the 180-day exclusivity period for ANDA filings. Eligibility for exclusivity would hinge on whether or not the applicant has entered into a disqualifying agreement. The bill has 6 sponsors from both parties. The bill has been added to the Eye on FDA tab for Proposed Legislation. (Note, this occurred earlier in March, but only caught my eye this week….)
Risk Communications in a Crowded World – Something I picked up from the advance Federal Register which seemed to garner a lot of interest when tweeted is the fact that FDA’s Risk Communications Advisory Committee is scheduling a meeting for April 29 and 30 at the White Oak Campus with the interesting agenda to “discuss general factors in risk communication about FDA-regulated products, including how to communicate effectively about FDA’s adverse event reporting systems, and messaging in the context of competing communicators.” FDA has been so slow to grapple with the brave new dimensions of communications, it should be an interesting meeting.
New Alerts Involving a Recall of Product Over Sterility Confidence – On March 21, FDA issued a new alert to providers and patients about a nationwide voluntary recall of all lots of sterile products that were produced and distributed by a single company in Georgia. The expanded recall came after reports of five patients diagnosed with serious eye infections. Earlier in the week, FDA announced a separate voluntary recall regarding a separate set of products from a New Jersey company.
FDA Approves New Magnetic Resonance Imaging Agent – The agency approved a new magnetic resonance imaging (MRI) product for use in imaging the brain, spine and associated tissues of patients aged 2 and older. According to the release from FDA, the product – Dotarem “helped radiologists better see CNS lesions” and “helped the radiologists identify lesion borders and other lesion features.” The product will carry a boxed warning about the risk of nephrogenic system fibrosis – a rare but serious condition associated with the use of gadolinium-based contrast agents such as Dotarem. Side effects, which were uncommon in clinical trials, included nausea, headache, pain or coldness at the injection site, and burning sensation. It is the seventh GBCA approved by FDA for patients undergoing CNS MRI.
That’s it for me this week. Have a good weekend folks!



{kind=link}